
Introduction
metabaR is an R package which supports the importing,
handling and post-bioinformatics evaluation and improvement of
metabarcoding data quality. It provides a suite of functions to identify
and filter common molecular artifacts produced during the experimental
workflow, from sampling to sequencing, ideally using experimental
controls.
Due to its simple structure, metabaR can easily be used
in combination with other R packages commonly used for ecological
analysis (vegan, ade4, ape,
picante, etc.). In addition, it provides flexible graphical
systems based on ggplot2 to vizualise data from both an
ecological and experimental perspective.
Dependencies and Installation
metabaR relies on basic R functions and data structures
so as to maximise fexibility and transposability across other packages.
It has a minimal number of dependencies to essential R packages :
-
ggplot2,cowplot, andigraphfor visualisation purposes -
reshape2for data manipulation purposes
-
veganandade4for basic data analyses
-
seqinrandbiomformatfor data import or formatting.
To install metabaR, use :
# install bioconductor dependencies
install.packages("BiocManager")
BiocManager::install("biomformat")
# install metabaR package
install.packages("remotes")
remotes::install_github("metabaRfactory/metabaR")And then load the package
Package overview
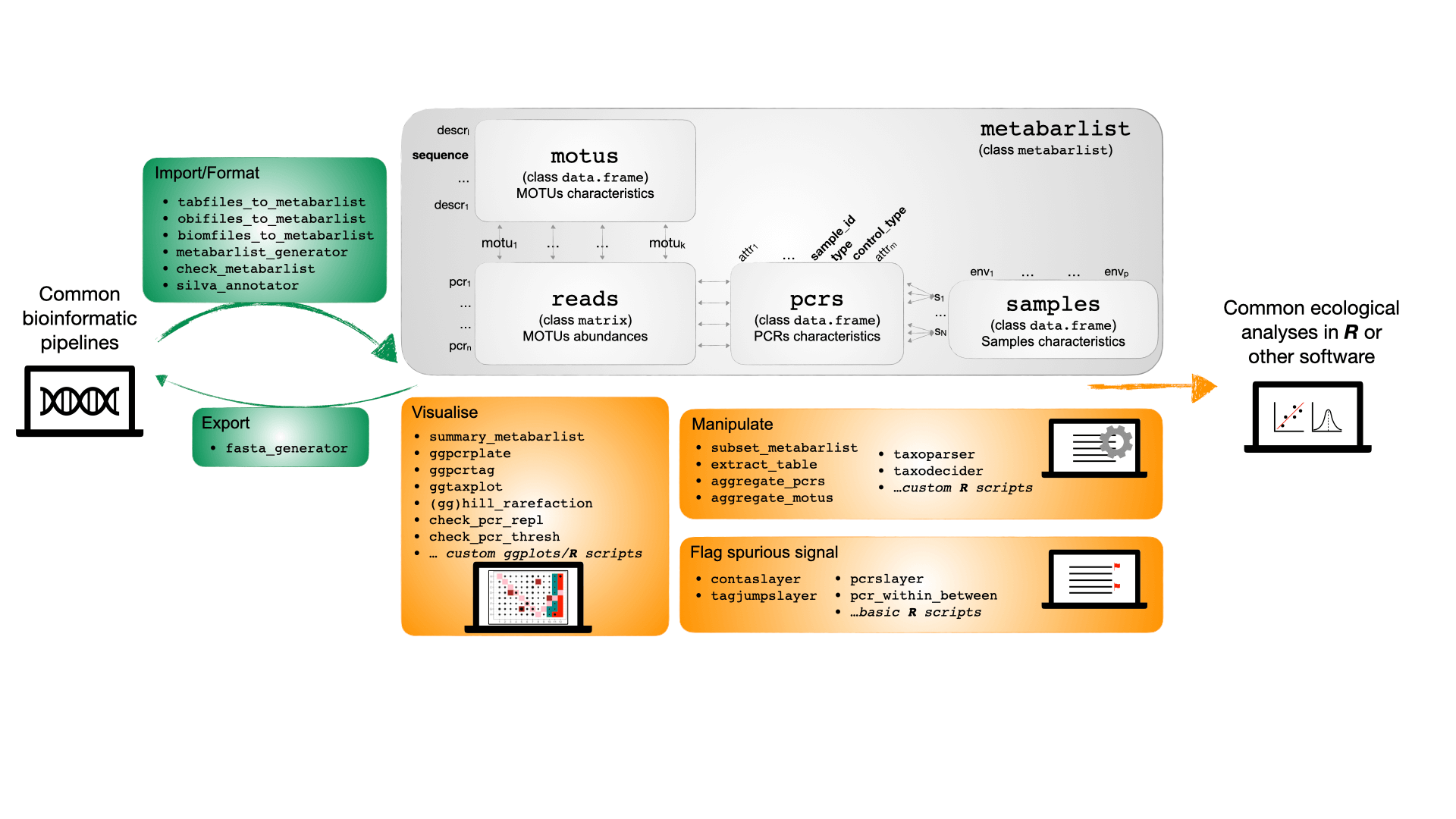
Data format and structure
The basic data format used in metabaR is a
metabarlist, a list of four tables:
readsa table of classmatrixconsisting of PCRs as rows, and molecular operational taxonomic units (MOTUs) as columns. The number of reads for each MOTU in each PCR is given in each cell, with 0 corresponding to no reads.motusa table of classdata.framewhere MOTUs are listed as rows, and their attributes as columns. A mandatory field in this table is the field “sequence”, i.e. the DNA sequence representative of the MOTU. Examples of other attributes that can be included in this table are the MOTU taxonomic information, the taxonomic assignment scores, MOTU sequence GC content, MOTU total abundance in the dataset, etc.-
pcrsa table of classdata.frameconsisting of PCRs as rows, and PCR attributes as columns. This table is particularly important inmetabaR, as it contains all the information related to the extraction, PCR and sequencing protocols and design that are necessary to assess and improve the quality of metabarcoding data (Taberlet et al. 2018; Zinger et al. 2019). This table can also include information relating to the PCR design, such as the well coordinates and plate of each PCR, the tag combinations specific of the PCR, the primers used, etc. Mandatory fields are:-
sample_id: a vector indicating the biological sample origin of each PCR (e.g. the sample name) -
type: the type of PCR, either a sample or an experimental control amplification. Only two values allowed:"sample"or"control".
-
control_type: the type of control. Only five values are possible in this field:-
NAiftype="sample", i.e. for any PCR obtained from a biological sample.
-
"extraction"for DNA extraction negative controls, i.e. PCR amplification of an extraction where the DNA template was replaced by extraction buffer.
-
"pcr"for PCR negative controls, i.e. pcr amplification where the DNA template was replaced by PCR buffer or sterile water.
-
"sequencing"for sequencing negative controls, i.e. unused tag/library index combinations.
-
"positive"for DNA extraction or PCR positive controls, i.e. pcr amplifications of known biological samples or DNA template (e.g. a mock community).
-
-
samplesa table of classdata.frameconsisting of biological samples as rows, and associated information as columns. Such information includes e.g. geographic coordinates, abiotic parameters, experimental treatment, etc. This table does not include information on the DNA metabarcoding experimental controls, which can only be found inpcrs.
Function Types
metabaR provides a range of function types:
- Import and formating functions to import DNA metabarcoding data from
common bioinformatic pipelines (e.g. OBITools, data in the biom format)
or more generally from any data formatted as described above in 4 tables
corresponding to the
reads,motus,pcrs,samples.
- Functions for data curation. These are often absent from most
bioinformatic pipelines. They aim to detect and enable the tagging of
potential molecular artifacts such as contaminants or failed PCR
reactions.
- Functions for visualizing the data under both ecological
(e.g. sample types similarity, rarefaction curves) and experimental
(e.g. control types, distribution across the PCR plate design)
perspectives.
- Functions to manipulate the
metabarlistobject, such as selection of a subset, data aggregation by PCRs or MOTUs, taxonomy information formatting, etc.
Example dataset
An example dataset is provided in metabaR to show how
the package can be used to assess and improve data quality.
The soil_euk dataset is of class
metabarlist. The data were obtained from an environmental
DNA (eDNA) metabarcoding experiment aiming to assess the diversity of
soil eukaryotes in French Guiana in two sites corresponding to two
contrasting habitats:
- Mana, a site located in a white sand forest, characterised by highly
oligotrophic soils and tree species adapted to the harsh local
conditions.
- Petit Plateau, a site located in the pristine rainforest of the
Nouragues natural reserve characterised by terra firme soils
richer in clay and organic matter.
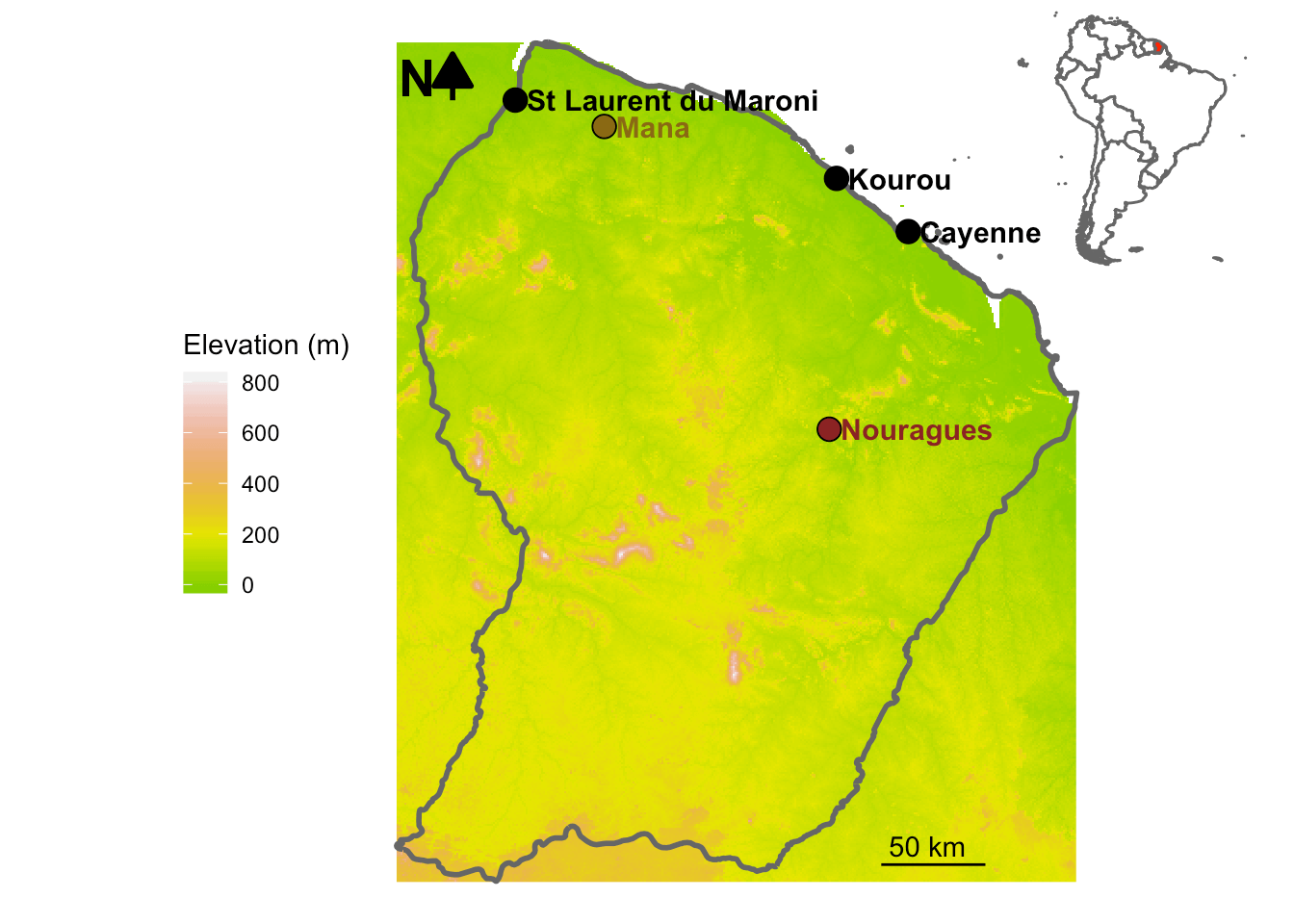
At each site, both soil and litter samples were collected so as to assess if these two types of material differ in their eukaryotic communities (Figure 2b). The total experiment consists of 384 PCR products, including 256 PCR products obtained from biological samples, the rest corresponding to various experimental controls. The retrieved data were then processed using the OBITools (Boyer et al. 2016) and SUMACLUST (Mercier et al. 2013) packages, as well as the SILVAngs pipeline (Quast et al. 2013).
More information on the soil_euk dataset can be found in
its help page:
?soil_eukThe example dataset is loaded in R as follows:
data(soil_euk)
summary_metabarlist(soil_euk)
#> $dataset_dimension
#> n_row n_col
#> reads 384 12647
#> motus 12647 15
#> pcrs 384 11
#> samples 64 8
#>
#> $dataset_statistics
#> nb_reads nb_motus avg_reads sd_reads avg_motus sd_motus
#> pcrs 3538913 12647 9215.919 10283.45 333.6849 295.440
#> samples 2797294 12382 10926.930 10346.66 489.5117 239.685The summary_metabarlist function displays the dataset
dimensions and characteristics. The above shows that the
soil_euk dataset is composed of 12647 eukaryote MOTUs from
384 pcrs, corresponding to a total of 64 soil cores plus the different
experimental controls (object soil_euk$dataset_dimension).
The total number of MOTUs and reads differ between the lines
pcrs and samples in the
soil_euk$dataset_statistics object because PCRs products
(hereafter referred to as pcrs) include experimental positive
and negative controls besides biological samples (hereafter referred to
as samples).
The soil_euk dataset also contains information relative
to MOTUs (15 variables in the motus table), PCRs (11
variables in the pcrs table), and samples (8 variables in
the samples table).
Such information can be observed with basic R commands, given that
the metabarlist object is a simple R list:
colnames(soil_euk$pcrs)
#> [1] "plate_no" "plate_col" "plate_row" "tag_fwd" "tag_rev"
#> [6] "primer_fwd" "primer_rev" "project" "sample_id" "type"
#> [11] "control_type"In soil_euk$pcrs these columns correspond to:
-
"plate_no": the PCR plate number in which each pcr has been conducted.
-
"plate_col"and"plate_row": the well coordinate (i.e. column and row) corresponding to where each pcr has been conducted.
-
"tag_fwd"and"tag_rev": the forward and reverse nucleotide tag/indices used to differenciate each pcr.
-
"primer_fwd"and"primer_rev": the forward and reverse primers used.
-
"project": the experiment project name. -
"sample_id","type","control_type": the mandatory fields for thepcrstable as described above.
colnames(soil_euk$samples)
#> [1] "site_id" "point_id" "Latitude"
#> [4] "Longitude" "Code.Petit.Plateau" "Site"
#> [7] "Habitat" "Material"In soil_euk$samples these columns correspond to:
-
"site_id"and"point_id": the identification codes for sites and sampling points respectively.
-
"Latitude"and"Longitude": the geographic coordinates of the sampling points (decimal degree).
-
"Code.Petit.Plateau": code specific to the experiment. Useful when several experiments are sequenced in the same library, but to not be considered in this tutorial.
-
"Site""Habitat": site name and corresponding habitat of each sample.
-
"Material": type of substrate material for each sample.
Tutorial with the soil_euk dataset
Below we provide a standard procedure for data analysis and curation
with the metabaR package. Note however that not all
functions provided in metabaR are indicated below, we
encourage the user to explore the package further in order to tailor its
use depending on your dataset characteristics.
Data import
metabaR provides import tools for different data
formats. Let’s consider for example a suite of four classical “.txt” or
csv files each corresponding to the future reads,
motus, pcrs, and samples objects.
These can be imported and formated into a metabarlist with
the tabfiles_to_metabarlist function as follows:
soil_euk <- tabfiles_to_metabarlist(file_reads = "litiere_euk_reads.txt",
file_motus = "litiere_euk_motus.txt",
file_pcrs = "litiere_euk_pcrs.txt",
file_samples = "litiere_euk_samples.txt")The default field separator of input files is tabulation, but this
can be modified by the user. The rows x columns of the input files
should follow the format of the table reads,
motus, pcrs, and samples
described above. Note that in this example above, these input files are
stored in the current working directory. If you want to use specifically
these example files, we refer you to the
tabfiles_to_metabarlist help page example.
Diagnostic Plots
Before processing the data, it is useful to begin the analysis with an explorative visualisation of the raw data, which can already highlight several potential problems.
Basic visualisation
An initial assessment consists in determining how many reads and
MOTUs have been obtained across samples and control types, with the
expectation that negative controls yield no or much lower read counts
and MOTU numbers. To do so, one can either create new independent
vectors storing the total number of reads and MOTUs in each
pcr, or store these information in the table pcrs
of the metabarlist directly, as done below:
# Compute the number of reads per pcr
soil_euk$pcrs$nb_reads <- rowSums(soil_euk$reads)
# Compute the number of motus per pcr
soil_euk$pcrs$nb_motus <- rowSums(soil_euk$reads>0)And then plot the results using the “control_type” column of the
pcrs table
# Load requested package for plotting
library(ggplot2)
library(reshape2)
# Create an input table (named check1) for ggplot of 3 columns:
# (i) control type
# (ii) a vector indicated whether it corresponds to nb_reads or nb_motus,
# (iii) the corresponding values.
check1 <- melt(soil_euk$pcrs[,c("control_type", "nb_reads", "nb_motus")])
ggplot(data <- check1, aes(x=control_type, y=value, color=control_type)) +
geom_boxplot() + theme_bw() +
geom_jitter(alpha=0.2) +
scale_color_manual(values = c("brown", "red", "cyan4","pink"), na.value = "darkgrey") +
facet_wrap(~variable, scales = "free_y") +
theme(axis.text.x = element_text(angle=45, h=1))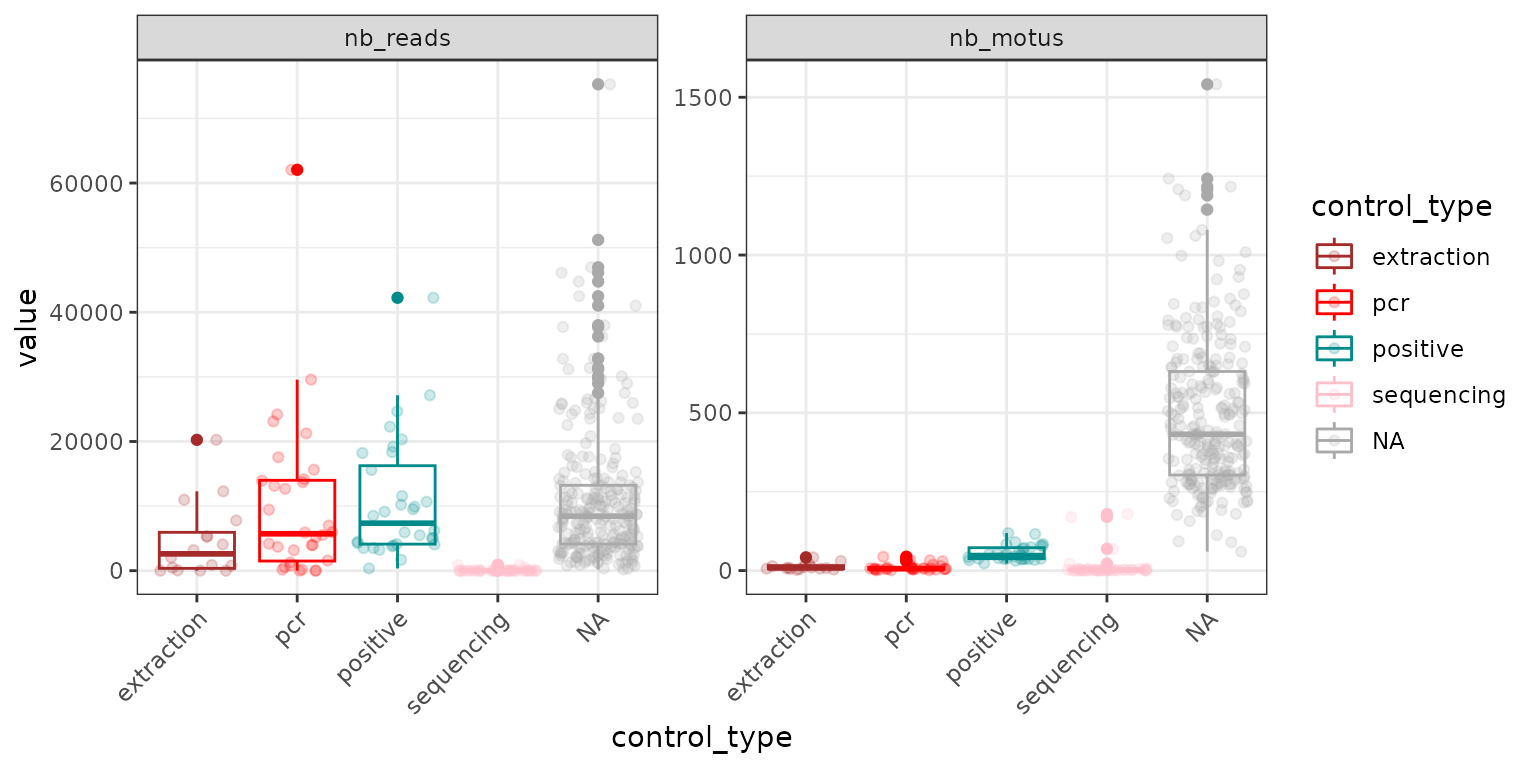
Remember that pcrs obtained from samples are
referred to as NA in the control_type vector,
shown in grey in the example above. Here, extraction and PCR negative
controls yield a few MOTUs of non negligible abundance, which are likely
contaminants. Not to worry for now, since this feature is common in DNA
metabarcoding datasets (Taberlet et al. 2018;
Zinger et al. 2019), and we will address with those below.
An other basic visualisation consists in determining how the number of MOTUs and reads correlate. This information can help identify the sequencing depth below which a pcr might not be reliable, in particular by comparison with the characteristics of experimental negative controls.
# Using the nb_reads and nb_motus defined previously in the soil_euk$pcrs table
ggplot(soil_euk$pcrs, aes(x=nb_reads, y=nb_motus, color = control_type)) +
geom_point() + theme_bw() +
scale_y_log10() + scale_x_log10() +
scale_color_manual(values = c("brown", "red", "cyan4","pink"), na.value = "darkgrey")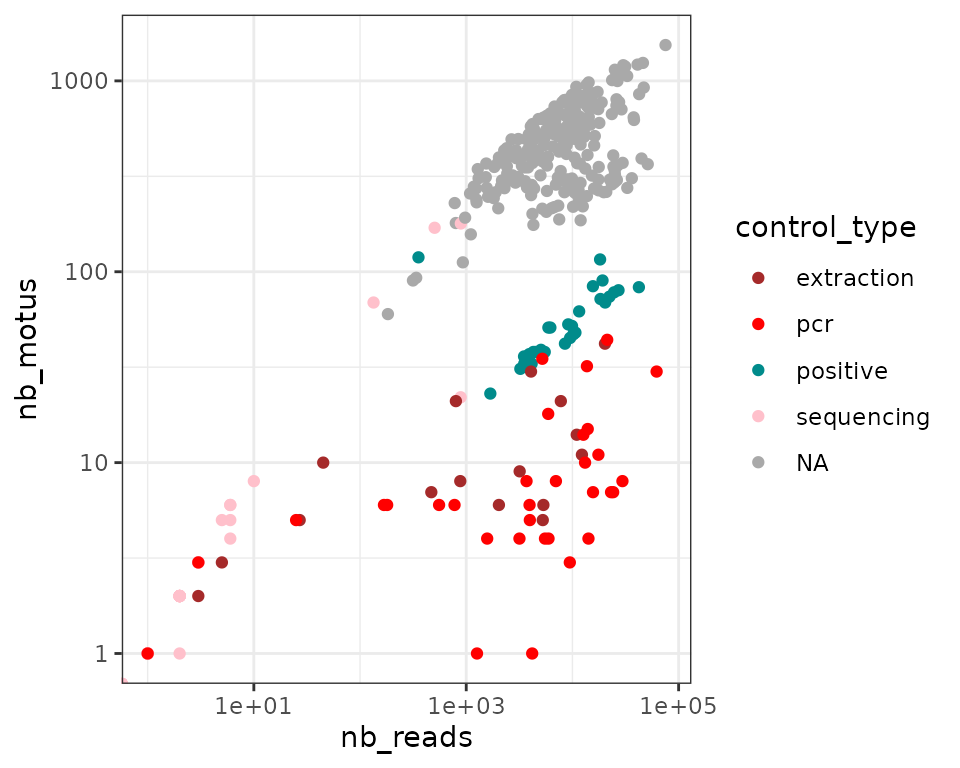
In general, we observe a positive correlation between the number of
reads and MOTUs per pcr. The strength of this relationship
for the different pcrs types (i.e. from samples or
controls) can reveal important information about the dataset. A strong
correlation suggests that the sequencing effort is not sufficient to
cover the pcr diversity. In other words, the number of MOTUs
increases linearly with the sequencing depth, which is not a desired
property when working with diversity. On the contrary, an absence of
correlation suggests that the diversity is well covered by sequencing.
The latter is the case in our example above with extraction or pcr
negative control pcrs harbouring high number of reads but low
numbers of MOTUs: these are most likely contaminants. The read / MOTU
number relationship is steepest for sequencing negative controls, which
indicates that although tag-jumps do occur at low rates in the
soil_euk dataset (low number of reads), it occurs for many
MOTUs (Schnell, Bohmann, and Gilbert
2015). For pcrs obtained from samples, the
number of reads and MOTUs is only slightly correlated. Of note is that
some pcrs exhibit features more similar to controls and are
likely failed.
Visualisation in the PCR design context
Another way to vizualise basic descriptive dataset statistics is to
represent them in their experimental context. For example, one can plot
the number of reads in the PCR plates if the PCR plate numbers and well
coordinates have been provided in the pcrs table. Such
visualisation can highlight potential issues at the PCR amplification
step. For example, low read abundances in pcrs throughout one
line or column of the PCR plate suggests that a primer was dysfunctional
or that the pipetting of reagents was inconsistent. Let’s see how it
appears for the soil_euk data:
ggpcrplate(soil_euk, FUN = function(m){rowSums(m$reads)}, legend_title = "# of reads per PCR")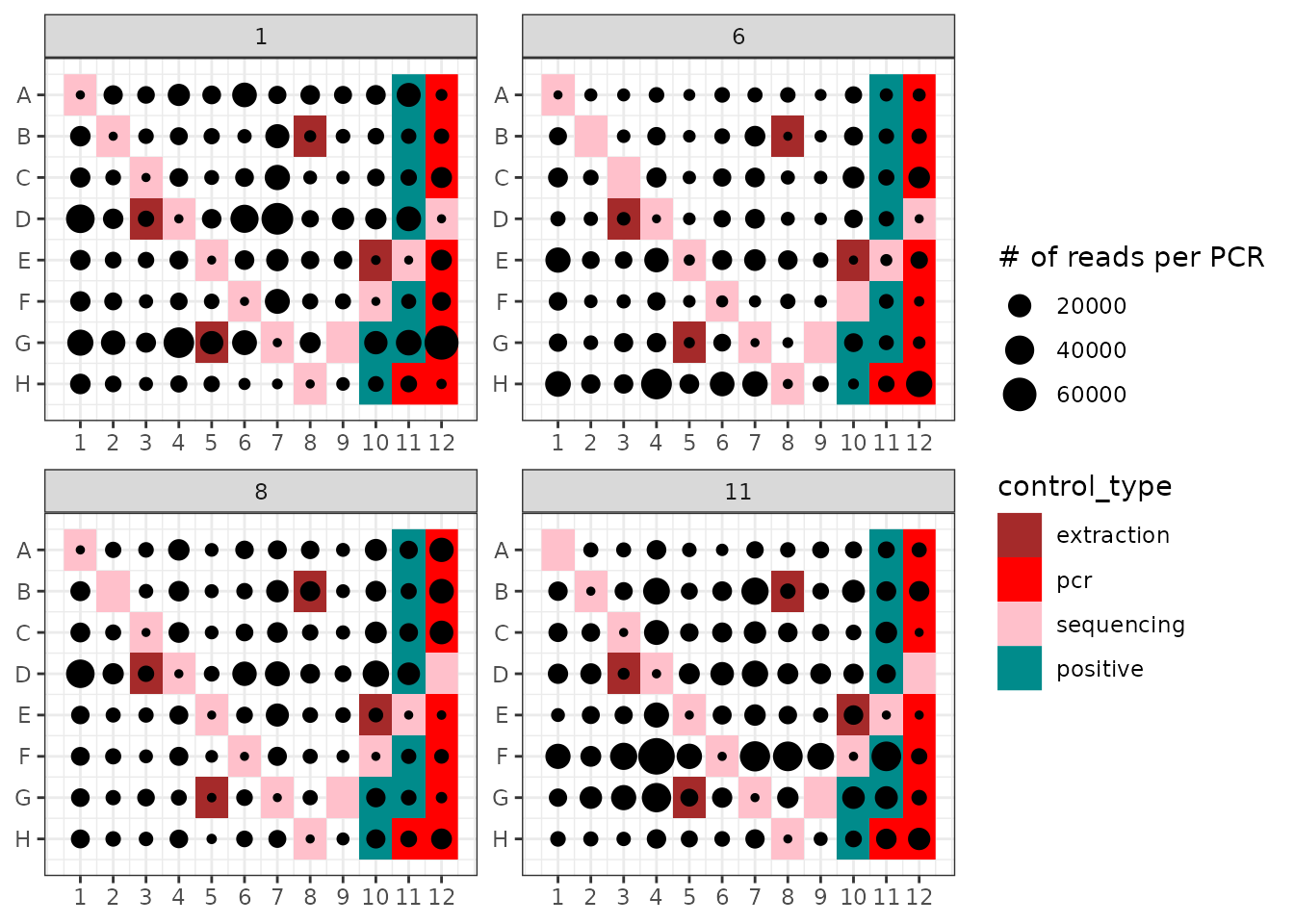
ggpcrplate uses a metabarlist and a custom
function. In the example above, it computes the number of reads per
pcr to display these in their experimental context. This
operation is the default when using ggpcrplate, and can be
obtained with the bfollowing asic call:
ggpcrplate(soil_euk)Besides the presence of contaminants in extraction and pcr negative
controls, one can observe:
- That sequencing negative controls (here corresponding to wells where
neither DNA template nor PCR reagents were introduced: these are the
unused tag combinations in the soil_euk dataset
experimental design), have low or null read numbers. This demonstrates
that although “tag-jumps” (Schnell, Bohmann, and
Gilbert 2015; reviewed in Zinger et al. 2019) are present, they
remain relatively limited in this experiment.
- That certain lines or columns do exhibit a low number of reads in
general, e.g. here in all wells in plate 1, line H. This tendency might
denote a pipetting or primer problem, as mentioned above.
If the PCR design uses a combination of two tags as for the
soil_euk dataset and that this information is available in
the pcrs table, it is also possible to determine if one of
the tag introduces systematic biases as follows:
# Here the list of all tag/indices used in the experiment
# is available in the column "tag_rev" of the soil_euk$pcrs table
tag.list <- as.character(unique(soil_euk$pcrs$tag_rev))
ggpcrtag(soil_euk,
legend_title = "# of reads per PCR",
FUN = function(m) {rowSums(m$reads)},
taglist = tag.list)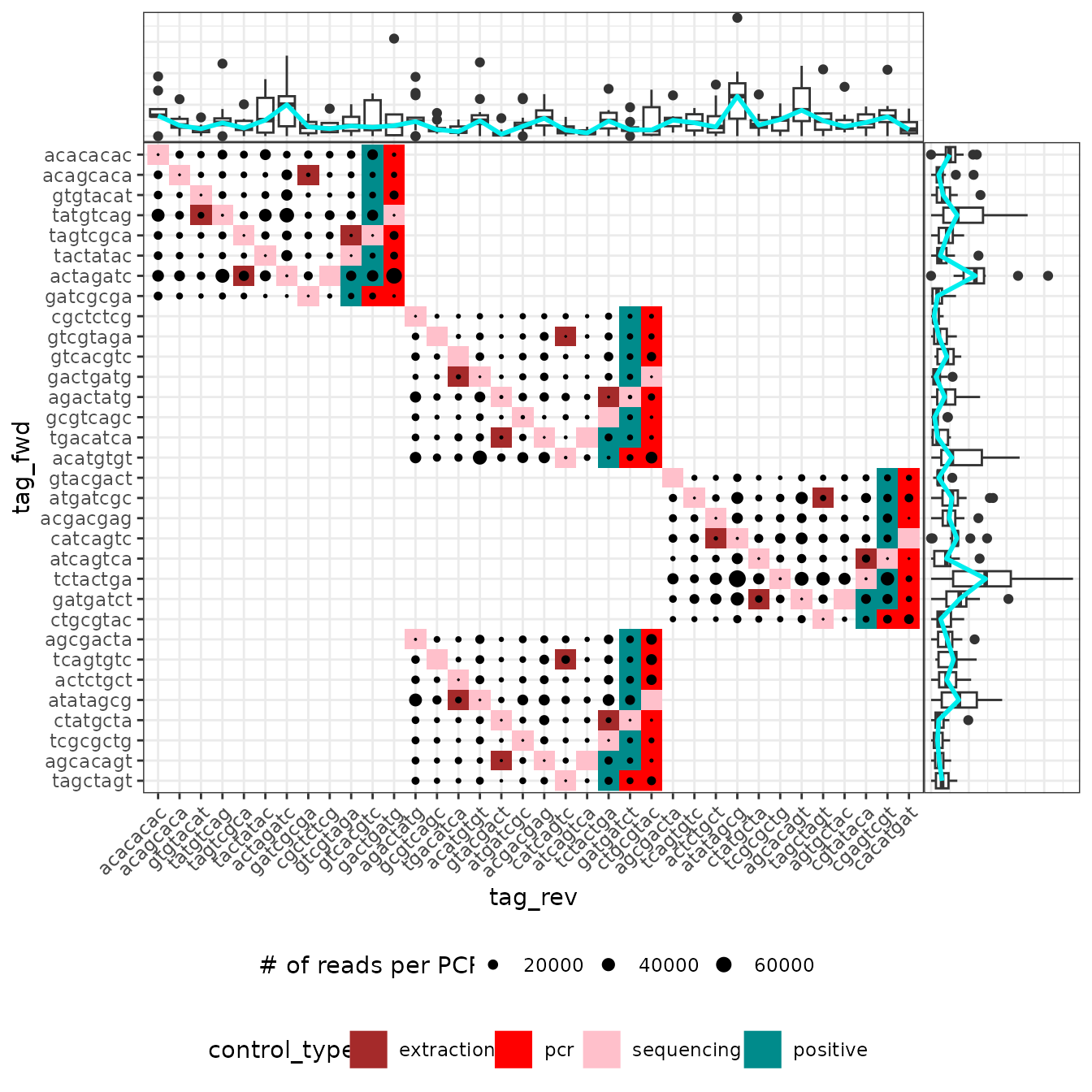
The ggpcrtag function works in a similar fashion to
ggpcrplate. The plot shows the number of reads in their
full PCR design, i.e. with all plates together including their shared
tags. The boxplots above and to the right show the distribution of the
number of reads obtained for each tag-primer. If the tagging strategy
relies on identical tags for both primers, then only the diagonal of
this plot will be shown.
Visualisation with tools borrowed from ecology
Another useful way to visualise the success of the experiment consists in constructing rarefaction curves, which can indicate whether sequencing effort is sufficient to cover the diversity in MOTUs of each pcr (remember here that sequencing is a sampling process of amplicons/MOTUs within the PCR tube, not of the diversity in the sampling site).
In metabaR, rarefaction curves are constructed with
three diversity indices. These are part of the Hill numbers framework
(reviewed in Chao, Chiu, and Jost 2014),
which is based on the concept of effective number species, and is
defined as follows:
Where
is the total number of species, and
the species frequency. The diversity
corresponds to the diversity of equally-common species, with order
defining the degree of species commonness. In other words, this
framework give less weight to rare species when
increases. The interesting feature of the framework is that it unifies
mathematically the best known diversity measures in ecology through this
unique parameter
:
for
,
is species richness
for
,
approaches the exponential of the Shannon index
for
,
is the inverted of the Simpson index
These are the value of
for which metabaR will construct rarefaction curves.
For metabarcoding data, these indices are particularly useful because they allow users to give less weight to rare MOTUs, which often correspond to molecular artifacts and of which inclusion in the data can lead to spurious ecological conclusions (Taberlet et al. 2018; Alberdi and Gilbert 2019; Calderón-Sanou et al. 2020).
metabaR also computes the Good’s coverage index for each
pcr:
Where is the number of singletons and the total number of individuals, i.e. the proportion of reads that are not singletons in the pcr. Note that the Good coverage index should be interpreted carefully with DNA metabarcoding data, as it is based on singletons. First, because singletons are potentially spurious signal that can be especially important is species- or DNA-poor samples. This may lead to an overestimation of the coverage. On the other hand, “absolute singletons” (i.e. singletons across the whole sequence dataset) are often filtered out during the bioinformatic process, so the number of singletons observed at this stage of the analysis is likely to be strongly underestimated (and this would artificially lower the coverage estimate).
Since this process can take a while to calculate with large datasets,
we here only construct rarefaction curves for a subset of pcrs,
in this case by keeping only few samples from the H20 plot of
the Petit Plateau. The subsetting of the soil_euk dataset
can be done as follows:
# Get the samples names from the H20 plot
h20_id <- grepl("H20-[A-B]", rownames(soil_euk$pcrs))
# Subset the data
soil_euk_h20 <- subset_metabarlist(soil_euk, table = "pcrs", indices = h20_id)
# Check results
summary_metabarlist(soil_euk_h20)
#> $dataset_dimension
#> n_row n_col
#> reads 16 3182
#> motus 3182 15
#> pcrs 16 13
#> samples 4 8
#>
#> $dataset_statistics
#> nb_reads nb_motus avg_reads sd_reads avg_motus sd_motus
#> pcrs 154553 3182 9659.562 7233.992 537.1875 157.0292
#> samples 154553 3182 9659.562 7233.992 537.1875 157.0292Note that subsetting of a metabarlist object can be done
with subset_metabarlist with any criterion, based either on
MOTUs, pcrs, or samples characteristics.
Now lets construct the rarefaction curves with the
hill_rarefaction function. The number of sequencing depths
used to build these curves is defined by the nsteps
argument. For each sequencing depth, the reads table is
resampled randomly nboot times so that to obtain an
estimate of
and
.
nboot is low in the example below to limit the computing
time.
soil_euk_h20.raref = hill_rarefaction(soil_euk_h20, nboot = 20, nsteps = 10)
head(soil_euk_h20.raref$hill_table)#> pcr_id reads D0 D0.sd D1 D1.sd D2 D2.sd
#> 1 H20-As_r1 5 4.60 0.598243 4.475125 1.180418 4.460317 0.7731693
#> 2 H20-As_r1 10 8.60 1.095445 8.150171 1.189881 7.901876 1.5261629
#> 3 H20-As_r1 100 52.20 3.592390 38.651053 1.098978 28.307113 3.4806900
#> 4 H20-As_r1 200 79.55 5.651968 48.041391 1.096751 30.467614 3.6207116
#> 5 H20-As_r1 500 137.45 7.308791 63.615132 1.065909 34.121794 2.6286914
#> 6 H20-As_r1 1000 201.90 8.931906 73.582977 1.057857 35.582662 1.7948423
#> coverage coverage.sd
#> 1 0.1600 0.239297217
#> 2 0.2600 0.181803827
#> 3 0.6590 0.051595593
#> 4 0.7510 0.034928498
#> 5 0.8489 0.017136910
#> 6 0.8966 0.008586403The hill_rarefaction function produces an object with
several elements. The most relevant one is the hill_table a
table indicating the pcr_id, the sequencing depth at which
the pcr was resampled, and the corresponding diversity /
coverage indices. The output of hill_rarefaction can be
used to draw rarefaction curves.
gghill_rarefaction(soil_euk_h20.raref)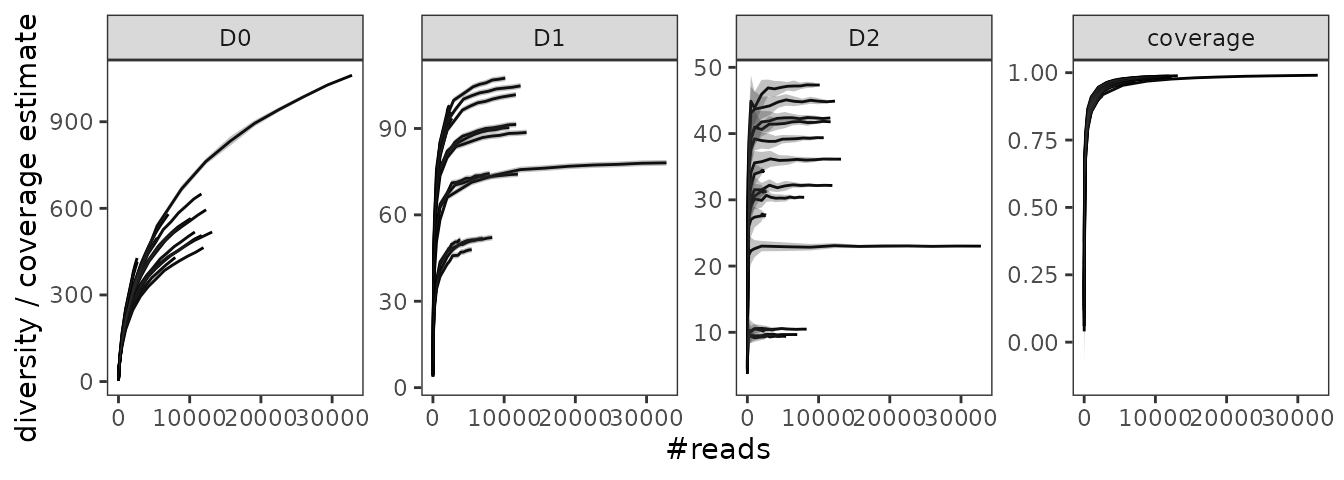
The user may also want to distinguish different types of samples on these curves, for example here soil vs. litter samples. This can be achieved as follows:
# Define a vector containing the Material info for each pcrs
material <- soil_euk_h20$samples$Material[match(soil_euk_h20$pcrs$sample_id,
rownames(soil_euk_h20$samples))]
# Use of gghill_rarefaction requires a vector with named pcrs
material <- setNames(material,rownames(soil_euk_h20$pcrs))
# Plot
p <- gghill_rarefaction(soil_euk_h20.raref, group=material)
p + scale_fill_manual(values = c("goldenrod4", "brown4", "grey")) +
scale_color_manual(values = c("goldenrod4", "brown4", "grey")) +
labs(color="Material type")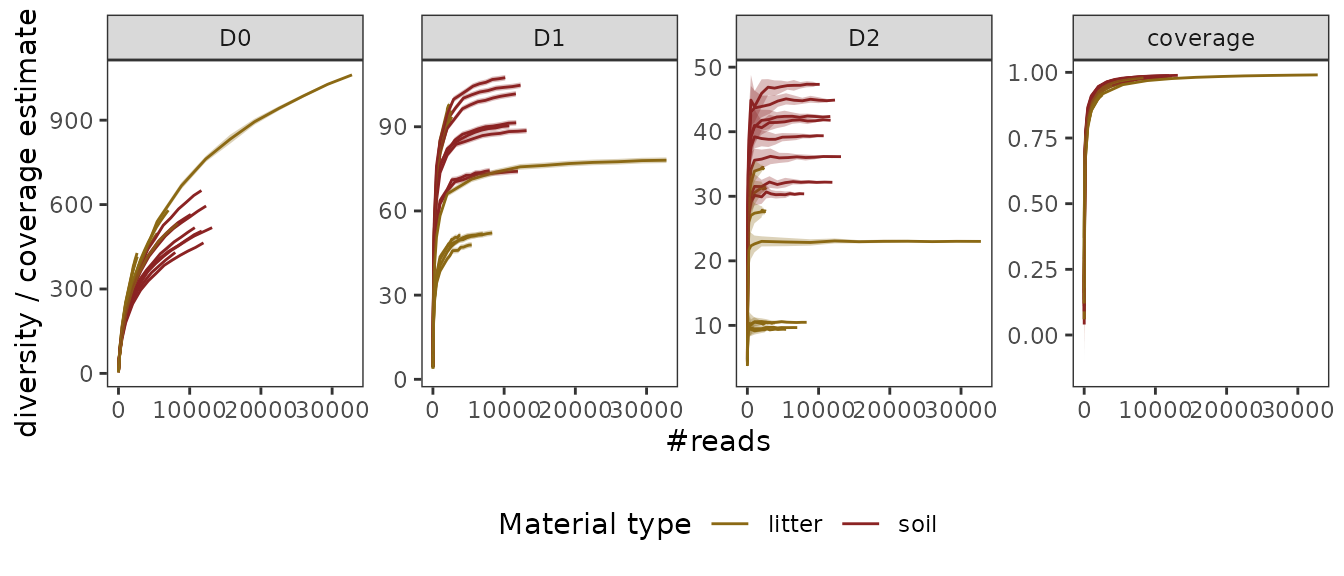
The obtained curves tell us a couple of things about the
pcrs:
- The sampling coverage is relatively high even at intermediate
sequencing depths (but see remarks above).
- The less weight is given to rare MOTUs (i.e. the higher
is), the better
is estimated.
- Litter samples in general tend to be less diverse than soil
samples.
Flagging spurious signal
The next steps of the analysis usually consist in identifying
potential spurious signal, whether in terms of MOTUs (e.g. contaminants,
untargeted taxa due to primer lack of specificity), MOTUs abundances
(i.e. tag-jumps ), or pcrs (pcrs yielding low amounts
of reads or with low replicability). In this tutorial, the flagging is
done by creating a boolean vector (i.e. TRUE/FALSE where
TRUE is good quality signal) specific to each flagging
criterion.
Detecting contaminants based on experimental negative controls
Contamination can occur at multiple stages, from sample collection to
library preparation. Detecting these contaminants can be done through
the use of negative controls at each stage (reviewed in Taberlet et al. 2018; Zinger et al.
2019), as is the case for the soil_euk dataset.
Due to the tagjump bias, many genuine MOTUs that are abundant in samples can be detected in negative controls. Consequently, simply removing from the dataset any MOTU that occurs in negative controls is a very bad idea.
A contaminant should be preferentially amplified in negative controls
since there is no competing DNA. The function contaslayer
relies on this assumption and detects MOTUs whose relative abundance
across the whole dataset is highest in negative controls. Note however
that this approach won’t be appropriate if the negative controls have
been contaminated with biological samples. In this
case,contaslayer should identify MOTUs that are dominants
in samples.
The function contaslayer adds a new column in table
motus indicating whether the MOTU is a genuine MOTU
(TRUE) or a contaminant (FALSE). Below, this
is demonstrated with the detection of contaminants from the DNA
extraction step.
# Identifying extraction contaminants
soil_euk <- contaslayer(soil_euk,
control_types = "extraction",
output_col = "not_an_extraction_conta")
table(soil_euk$motus$not_an_extraction_conta)
#>
#> FALSE TRUE
#> 66 12581contaslayer identified here 66 contaminant MOTUs. Below
are the ten most common extraction contaminants identified by
contaslayer in the soil_euk dataset.
| total # reads | similarity to ref DB | full taxonomic path | sequence |
|---|---|---|---|
| 195920 | 1 | root, Eukaryota | ctcaaacttccatcggcttgagccgatagtccctctaagaagccggcgaccagccaaagctagcctggctatttagcaggttaaggtctcgttcgttat |
| 23499 | 1 | root, Eukaryota, Viridiplantae, Streptophyta, Streptophytina, Embryophyta, Tracheophyta, Euphyllophyta, Spermatophyta, Magnoliophyta, Mesangiospermae, eudicotyledons, Gunneridae, Pentapetalae, rosids, malvids, Sapindales | ctcaaacttccttggcctaagcggccatagtccctctaagaagctggccacggagggatacctccgcatagctagttagcaggctgaggtctcgttcgttaa |
| 8296 | 1 | root, Eukaryota, Opisthokonta, Fungi, Dikarya, Ascomycota, saccharomyceta, Saccharomycotina, Saccharomycetes, Saccharomycetales | ctcaaacttccatcgacttgaaatcgatagtccctctaagaagtgactataccagcaaaagctagcagcactatttagtaggttaaggtctcgttcgttat |
| 4010 | 1 | root | ctcaaacttccatcggcttgaaaccgatagtccctctaagaagtggataaccagcaaatgctagcaccactatttagtaggttaaggtctcgttcgttat |
| 1055 | 1 | root, Eukaryota, Opisthokonta, Fungi, Dikarya, Basidiomycota, Ustilaginomycotina, Exobasidiomycetes, Entylomatales | ctcaaacttccattagctaaacgccaatagtccctctaagaagccagcggctaaccatagtcggccgggctatttagcaggttaaggtctcgttcgttat |
| 966 | 1 | root, Eukaryota, Stramenopiles, Chrysophyceae, Chromulinales, Chromulinaceae | ccccaacttcctttggttagtcaccaaaagtccctctaagaagcttacgtcaatactagtgcattaacaaaactatttagcaggcgggggtctcgttcgttaa |
| 611 | 1 | root, Eukaryota, Opisthokonta, Fungi, Dikarya, Basidiomycota, Agaricomycotina, Tremellomycetes | ctcaaacttccaacagctaaacgctgttagtccctctaagaagacagcggccagcaaaagccgaccggtctatttagcaggttaaggtctcgttcgttat |
| 473 | 1 | root, Eukaryota, Viridiplantae, Streptophyta, Streptophytina, Embryophyta, Tracheophyta, Euphyllophyta, Spermatophyta, Magnoliophyta, Mesangiospermae, Liliopsida, Petrosaviidae, commelinids, Poales, Poaceae, BOP clade, Pooideae | ctcaaacttccgtcgcctaaacggcgatagtccctctaagaagctagctgcggagggatggctccgcatagctagttagcaggctgaggtctcgttcgttaa |
| 456 | 1 | root, Eukaryota, Opisthokonta, Fungi, Dikarya, Basidiomycota, Ustilaginomycotina, Malasseziomycetes, Malasseziales, Malasseziaceae, Malassezia | ctcaaacttccattggctaaacgccaatagtccctctaagaagccagcagccaaccatagtcgactgggctatttagcaggttaaggtctcgttcgttat |
| 189 | 1 | root, Eukaryota, Viridiplantae, Streptophyta, Streptophytina, Embryophyta, Tracheophyta, Euphyllophyta, Spermatophyta, Magnoliophyta, Mesangiospermae | ctcaaacttccgtggcctaaacggccatagtccctctaagaagctggccgcggagggatgcctccgtgtagctagttagcaggctgaggtctcgttcgttat |
The identified contaminants correspond to metazoans (including
humans), protists, fungi and plants. The most abundant contaminant does
not have fine taxonomic identification in the soil_euk
dataset, but a BLAST search of the sequence in NCBI suggests that it
most likely corresponds to a Fusarium, a notorious laboratory
contaminant.
Next, one may want to know how these contamiants are distributed
across the soil_euk dataset. For example, the contamination
could be present only in one PCR plate for some reason. To assess this,
one could use ggpcrplate to plot the distribution of the
most common contaminant across the PCR setup:
# Identify the most common contaminant
# get contaminant ids
id <- !soil_euk$motus$not_an_extraction_conta
max.conta <- rownames(soil_euk$motus[id,])[which.max(soil_euk$motus[id, "count"])]
#... and its distribution and relative abundance in each pcr
ggpcrplate(soil_euk, legend_title = "#reads of most \nabundant contaminant",
FUN = function(m) {m$reads[, max.conta]/rowSums(m$reads)})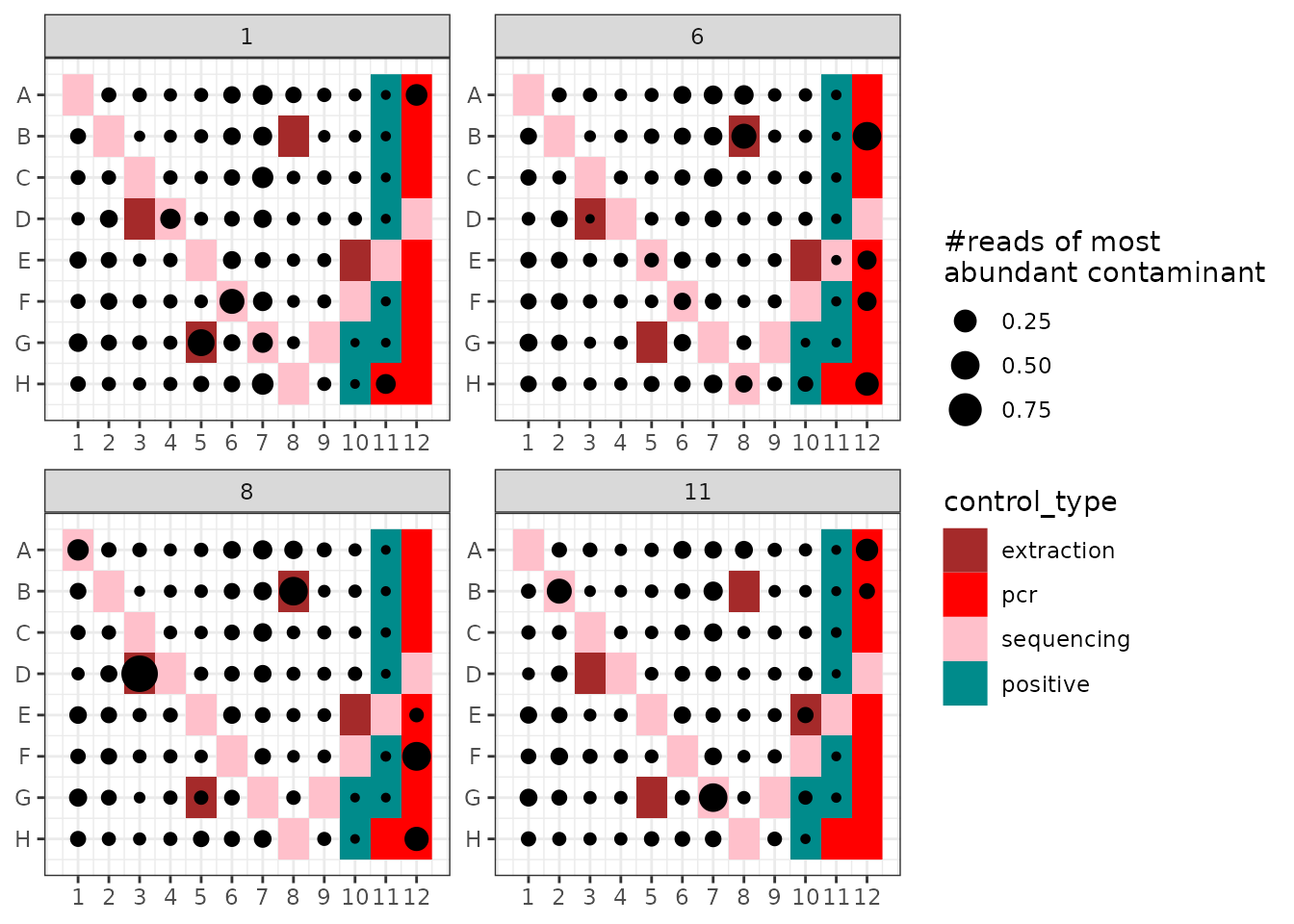
In the soil_euk dataset, the most abundant contaminant
occurs across all samples, but in general at low relative abundance (on
average 1.82 %) here.
One can also determine whether pcrs exhibit generally high proportions of contaminants.
# Compute relative abundance of all pcr contaminants together
a <- data.frame(conta.relab = rowSums(soil_euk$reads[,!soil_euk$motus$not_an_extraction_conta]) /
rowSums(soil_euk$reads))
# Add information on control types
a$control_type <- soil_euk$pcrs$control_type[match(rownames(a), rownames(soil_euk$pcrs))]
ggplot(a, aes(x=control_type, y=conta.relab, color=control_type)) +
geom_boxplot() + geom_jitter(alpha=0.5) +
scale_color_manual(values = c("brown", "red", "cyan4","pink"), na.value = "darkgrey") +
labs(x=NULL, y="Prop. Reads (log10)") +
theme_bw() +
scale_y_log10()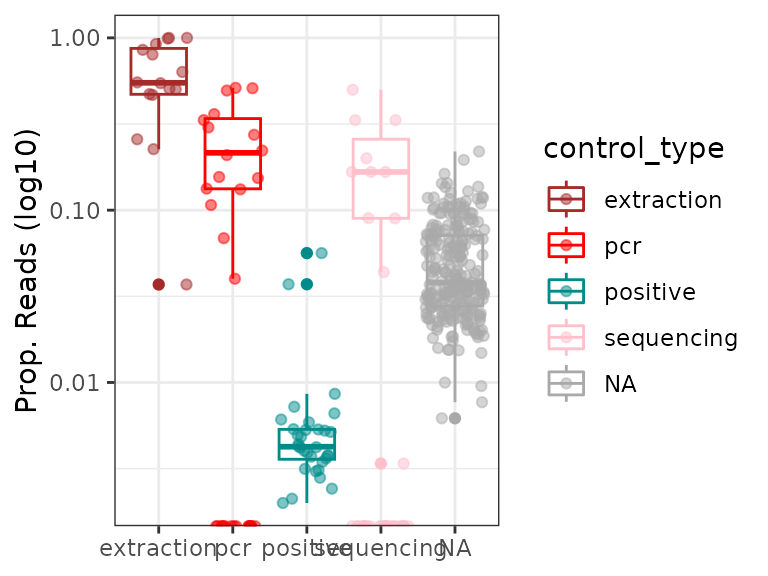
Overall, samples yield much less amounts of extraction contaminants than experimental negative controls. Some pcrs corresponding to samples have, however, > 10% of their reads corresponding to contaminants and could be unusable. Based on this criteria pcrs could be flagged as follows for potential downstream filtering:
# flag pcrs with total contaminant relative abundance > 10% of reads)
soil_euk$pcrs$low_contamination_level <-
ifelse(a$conta.relab[match(rownames(soil_euk$pcrs), rownames(a))]>1e-1, F, T)
# Proportion of potentially functional (TRUE) vs. failed (FALSE) pcrs
# (controls included) based on this criterion
table(soil_euk$pcrs$low_contamination_level) / nrow(soil_euk$pcrs)
#>
#> FALSE TRUE
#> 0.1666667 0.8046875Note that this step should be done for all types of contaminants (coming from extraction, pcr or sequencing). The user can identify each of them separetly or without differentiating the different controls types.
Flagging spurious or non-target MOTUs
Non-target sequences can be amplified if the primers are not specific enough. On the other hand, some highly degraded sequences can be produced throughout the data production process, such as primer dimers, or chimeras from multiple parents (hereafter referred to as spurious MOTUs). To detect these, one can use the information related to taxonomic assignments and associated similarity scores.
Since the soil_euk dataset was obtained with primers
that target eukaryotes, non-eukaryotic MOTUs should be excluded. At this
stage of the analysis, we only flag MOTUs based on this criterion.
#Flag MOTUs corresponding to target (TRUE) vs. non-target (FALSE) taxa
soil_euk$motus$target_taxon <- grepl("Eukaryota", soil_euk$motus$path)
# Proportion of each of these over total number of MOTUs
table(soil_euk$motus$target_taxon) / nrow(soil_euk$motus)
#>
#> FALSE TRUE
#> 0.04356764 0.95643236
# Intersection with extraction contaminant flags (not contaminant = T)
table(soil_euk$motus$target_taxon,
soil_euk$motus$not_an_extraction_conta)
#>
#> FALSE TRUE
#> FALSE 8 543
#> TRUE 58 12038Here we flagged 8 MOTUS as non-targets, which were already flagged as potential extraction contaminants.
Next, we want to identify MOTUs whose sequence is too dissimilar from references. This filtering criterion relies on the assumption that current reference databases capture most of the diversity at broad taxonomic levels (i.e. already have for example at least one representative of each phyla). Considering this, MOTUs being too distant from reference databases are more likely to be a degraded sequence, especially if such MOTUs are relatively numerous and of low abundance. To assess this, one can use the distribution of MOTU similarity scores, weighted and unweighted by their relative abundance.
# Plot the unweighted distribution of MOTUs similarity scores
a <-
ggplot(soil_euk$motus, aes(x=best_identity.order_filtered_embl_r136_noenv_EUK)) +
geom_histogram(color="grey", fill="white", bins=20) +
geom_vline(xintercept = 0.8, col="orange", lty=2) +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(x="% similarity against best match", y="# MOTUs")
# Same for the weighted distribution
b <-
ggplot(soil_euk$motus,
aes(x=best_identity.order_filtered_embl_r136_noenv_EUK, y = ..count.., weight = count)) +
geom_histogram(color="grey", fill="white", bins=20) +
geom_vline(xintercept = 0.8, col="orange", lty=2) +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(x="% similarity against best match", y="# Reads")
# Combine plots into one
library(cowplot)
ggdraw() +
draw_plot(a, x=0, y=0, width = 0.5) +
draw_plot(b, x=0.5, y=0, width = 0.5)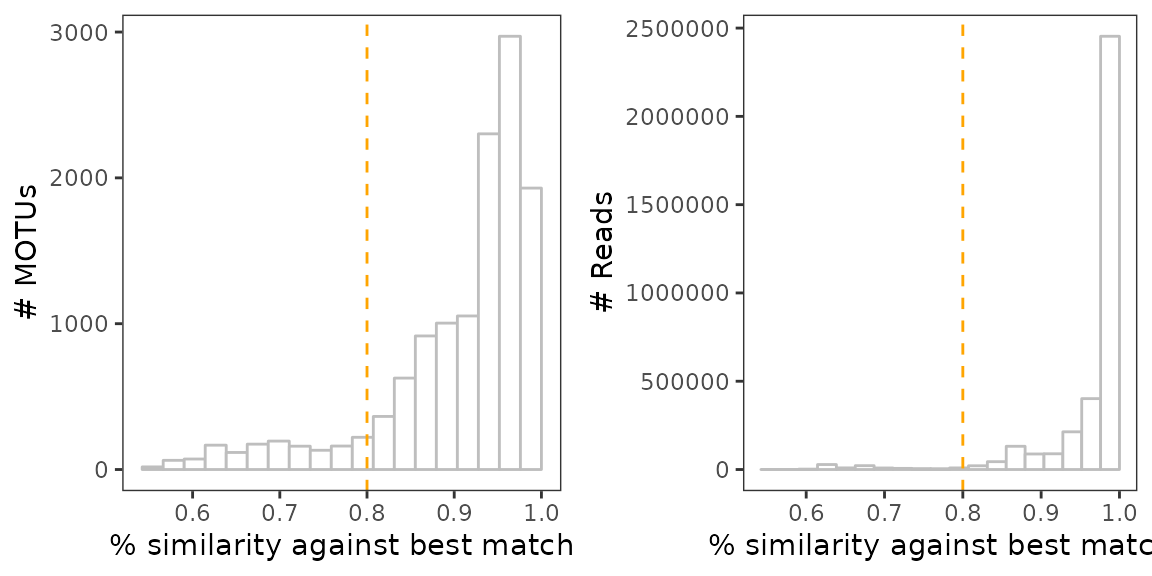
Here we may consider any MOTU as degraded sequences if its sequence similarity is < 80% against its best match in the reference database.
# Flag not degraded (TRUE) vs. potentially degraded sequences (FALSE)
soil_euk$motus$not_degraded <-
ifelse(soil_euk$motus$best_identity.order_filtered_embl_r136_noenv_EUK < 0.8, F, T)
# Proportion of each of these over total number of MOTUs
table(soil_euk$motus$not_degraded) / nrow(soil_euk$motus)
#>
#> FALSE TRUE
#> 0.111568 0.888432
# Intersection with other flags
table(soil_euk$motus$target_taxon,
soil_euk$motus$not_an_extraction_conta,
soil_euk$motus$not_degraded)
#> , , = FALSE
#>
#>
#> FALSE TRUE
#> FALSE 7 438
#> TRUE 10 956
#>
#> , , = TRUE
#>
#>
#> FALSE TRUE
#> FALSE 1 105
#> TRUE 48 11082Here, we find that 7 non-target MOTUs are also extraction contaminants and potentially degraded fragments.
Detecting PCR outliers
A first way to identify failed PCRs is to flag them based on the pcr sequencing depth
ggplot(soil_euk$pcrs, aes(nb_reads)) +
geom_histogram(bins=40, color="grey", fill="white") +
geom_vline(xintercept = 1e3, lty=2, color="orange") + # threshold
scale_x_log10() +
labs(x="# Reads (with all MOTUs and PCRs)",
y="# PCRs") +
theme_bw() +
theme(panel.grid = element_blank())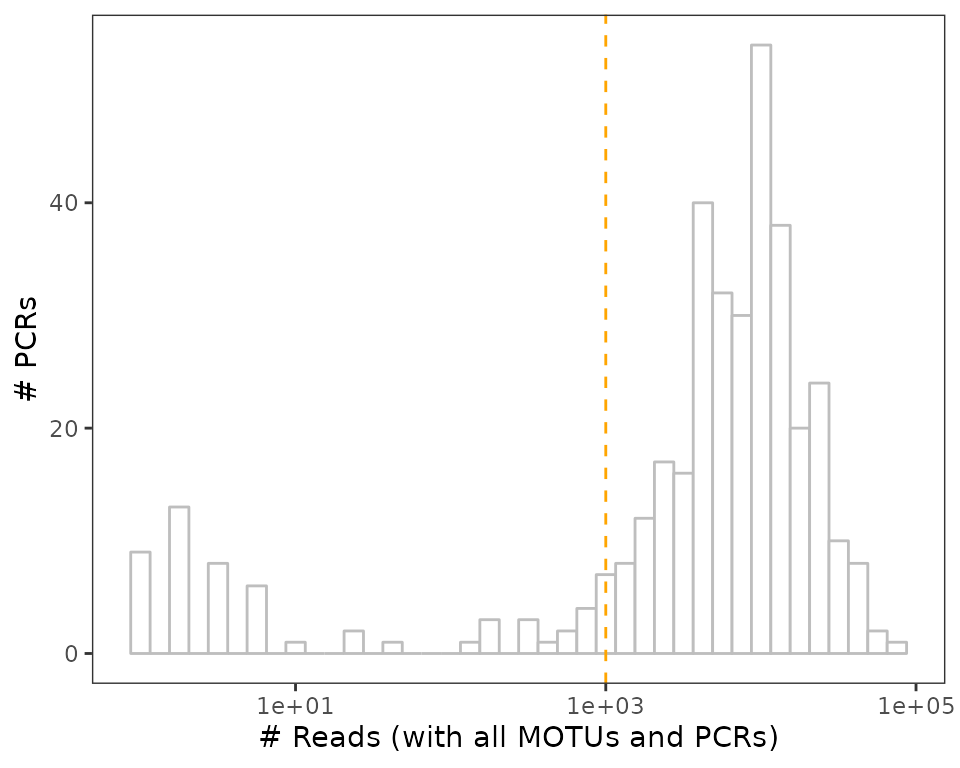
The histogram above includes negative controls, which - fortunately - yield low amounts of reads in general, but also pcrs obtained from samples, as shown below.
# Flag pcrs with an acceptable sequencing depth (TRUE) or inacceptable one (FALSE)
soil_euk$pcrs$seqdepth_ok <- ifelse(soil_euk$pcrs$nb_reads < 1e3, F, T)
# Proportion of each of these over total number of pcrs, control excluded
table(soil_euk$pcrs$seqdepth_ok[soil_euk$pcrs$type=="sample"]) /
nrow(soil_euk$pcrs[soil_euk$pcrs$type=="sample",])
#>
#> FALSE TRUE
#> 0.02734375 0.97265625A second way to evaluate the PCR quality is to assess their
reproducibility. The pcrslayer and associated functions
(e.g. pcr_within_between) enable the detection of outlier
replicates within pcrs using different methods, with a common
underlying assumption: the compositional dissimilarities in MOTUs
between replicate pcrs from a same sample should be
lower than that observed between pcrs obtained from different
samples (see help page for more details). This assessment is
obviously only possible when the experimental designs includes PCR
replicates for each biological samples. For these function to run, pcrs
yielding no reads, as well as negative controls, should be excluded.
# Subsetting the metabarlist
soil_euk_sub <- subset_metabarlist(soil_euk,
table = "pcrs",
indices = soil_euk$pcrs$nb_reads>0 & (
is.na(soil_euk$pcrs$control_type) |
soil_euk$pcrs$control_type=="positive"))
# First visualization
comp1 = pcr_within_between(soil_euk_sub)
check_pcr_thresh(comp1)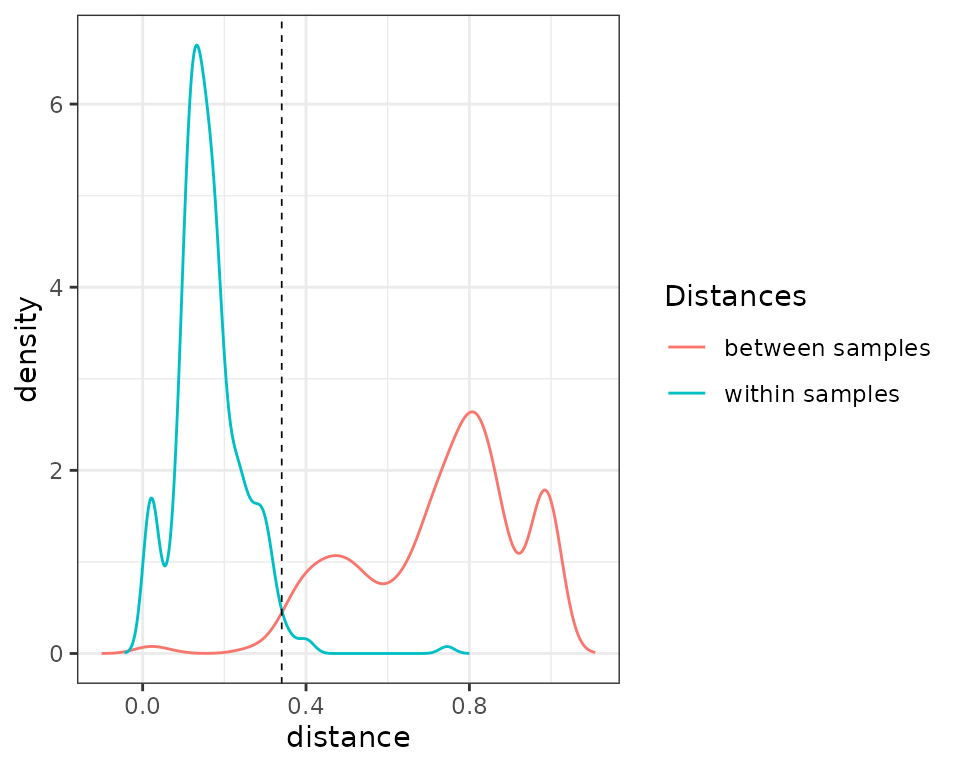
In the density plots shown above, one can see that several
pcrs replicates are as distant than pcrs from
different samples. Let’s now flag pcrs based on this
criterion and store this in the output_col column of the
pcr table:
# Flagging
soil_euk_sub <- pcrslayer(soil_euk_sub, output_col = "replicating_pcr", plot = F)
#> [1] "Iteration 1"
#> [1] "Iteration 2"
#> [1] "Iteration 3"
# Proportion of replicating pcrs (TRUE)
table(soil_euk_sub$pcrs$replicating_pcr) /
nrow(soil_euk_sub$pcrs)
#>
#> FALSE TRUE
#> 0.01736111 0.98263889
# Intersection with the sequencing depth criterion
table(soil_euk_sub$pcrs$seqdepth_ok,
soil_euk_sub$pcrs$replicating_pcr)
#>
#> FALSE TRUE
#> FALSE 3 5
#> TRUE 2 278Here, 3 pcrs out of the 5 detected as outliers had low
sequencing depth. One can also visualise the replicates and outliers
behaviour in a Principal Coordinate Analysis with the function
check_pcr_repl.
# Distinguish between pcrs obtained from samples from positive controls
mds = check_pcr_repl(soil_euk_sub,
groups = soil_euk_sub$pcrs$type,
funcpcr = soil_euk_sub$pcrs$replicating_pcr)
mds + labs(color = "pcr type") + scale_color_manual(values = c("cyan4", "gray"))Now report the flagging in the initial metabarlist
soil_euk$pcrs$replicating_pcr <- NA
soil_euk$pcrs[rownames(soil_euk_sub$pcrs),"replicating_pcr"] <- soil_euk_sub$pcrs$replicating_pcrLowering tag-jumps
Tag-jumps are frequency-dependent, i.e. abundant genuine MOTUs are
more likely to be found in low abundance in samples were they are not
supposed to be than rare genuine MOTUs. To reduce the amount of such
false positives, the function tagjumpslayer considers each
MOTU separately and corrects its abundance in pcrs (see
tagjumpslayer help for more information on possible
correction methods) when the MOTU relative abundance over the entire
dataset is below a given threshold. Such data a curation strategy is
similar to what has been proposed by Esling,
Lejzerowicz, and Pawlowski (2015). Effect of this threshold can
be evaluated by testing how this filtration procedure affects basic
dataset characteristics (e.g. # MOTUs or reads) at different levels, as
exemplified below.
# Define a vector of thresholds to test
thresholds <- c(0,1e-4,1e-3, 1e-2, 3e-2, 5e-2)
# Run the tests and stores the results in a list
tests <- lapply(thresholds, function(x) tagjumpslayer(soil_euk,x))
names(tests) <- paste("t_", thresholds, sep="")
# Format the data for ggplot with amount of reads at each threshold
tmp <- melt(as.matrix(do.call("rbind", lapply(tests, function(x) rowSums(x$reads)))))
colnames(tmp) <- c("threshold", "sample", "abundance")
# Add richness in MOTUs at each threshold
tmp$richness <-
melt(as.matrix(do.call("rbind", lapply(tests, function(x) {
rowSums(x$reads > 0)
}))))$value
# Add control type information on pcrs and make data curation threshold numeric
tmp$controls <- soil_euk$pcrs$control_type[match(tmp$sample, rownames(soil_euk$pcrs))]
tmp$threshold <- as.numeric(gsub("t_", "", tmp$threshold))
# New table formatting for ggplot
tmp2 <- melt(tmp, id.vars=colnames(tmp)[-grep("abundance|richness", colnames(tmp))])
ggplot(tmp2, aes(x=as.factor(threshold), y=value)) +
geom_boxplot(color="grey40") +
geom_vline(xintercept = which(levels(as.factor(tmp2$threshold)) == "0.01"), col="orange", lty=2) +
geom_jitter(aes(color=controls), width = 0.2, alpha=0.5) +
scale_color_manual(values = c("brown", "red", "cyan4","pink"), na.value = "darkgrey") +
facet_wrap(~variable+controls, scale="free_y", ncol=5) +
theme_bw() +
scale_y_log10() +
labs(x="MOTU pcr : total abundance filtering threshold", y="# Reads/MOTUs") +
theme(panel.grid = element_blank(),
strip.background = element_blank(),
axis.text.x = element_text(angle=40, h=1),
legend.position = "none")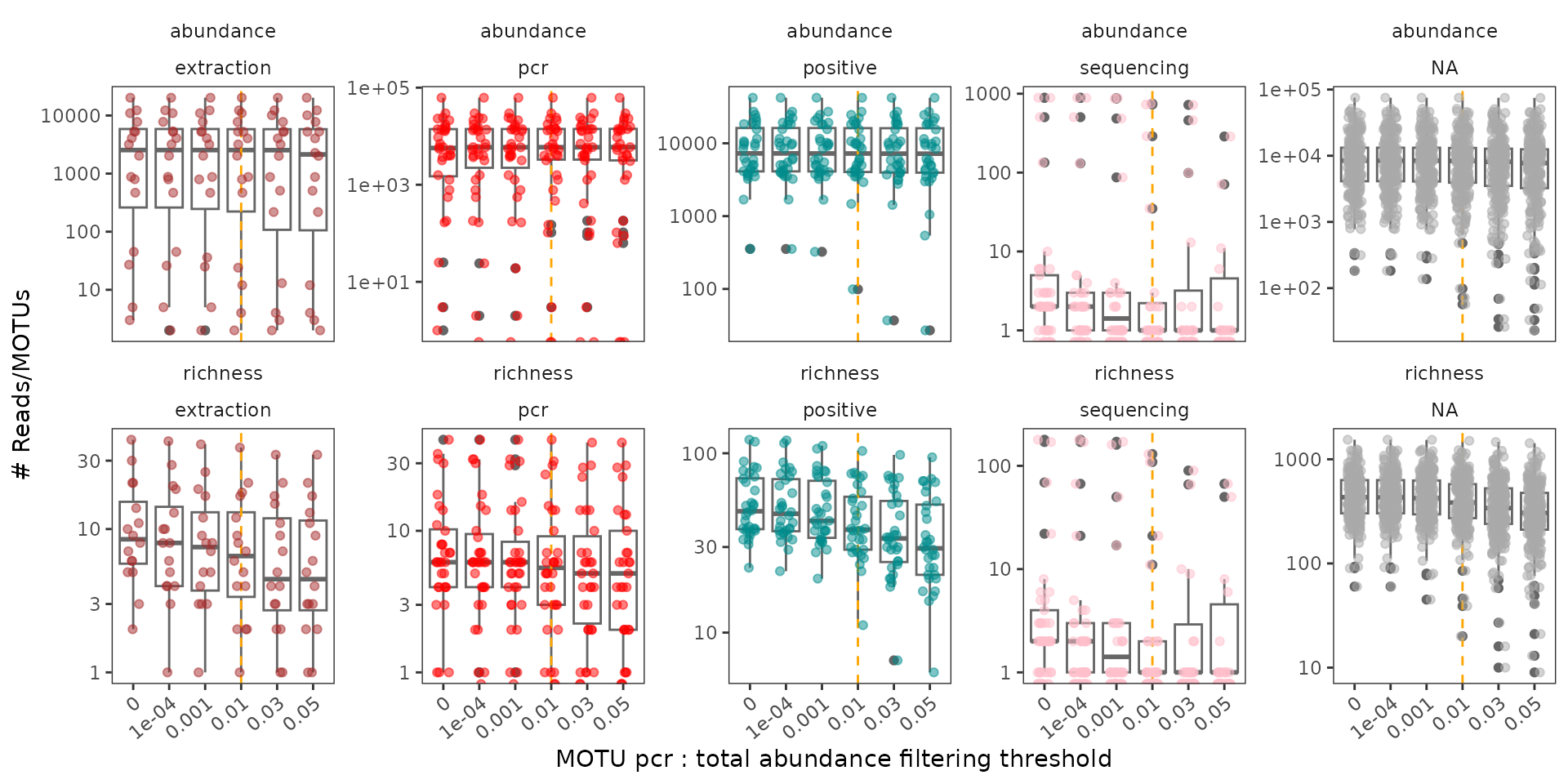
A threshold of 0.01 leads to a drop in both the number of reads and of MOTUs in sequencing negative controls. This drop is also noticeable in terms of the number of MOTUs in pcrs obtained from other controls as compared to those obtained from samples. The former are expected to be void of environmental MOTUs, and tag-jumps should be more visible/important in these pcrs. Note that this procedure primarily affects MOTU diversity in pcrs, and poorly the number of reads in pcrs.
As for above, pcrs containing large amounts of MOTUs identified as potentially artifactual or where tag-jumps filtering strongly affects the number of reads in pcrs can be flagged as potentially failed.
Summarizing the noise in the soil_euk dataset
We can now get an overview of the amount of noise identified with the criteria used above, for both the number of MOTUs and their associated readcount.
# Create a table of MOTUs quality criteria
# noise is identified as FALSE in soil_euk, the "!" transforms it to TRUE
motus.qual <- !soil_euk$motus[,c("not_an_extraction_conta", "target_taxon", "not_degraded")]
colnames(motus.qual) <- c("extraction_conta", "untargeted_taxon", "degraded_seq")
# Proportion of MOTUs potentially artifactual (TRUE) based on the criteria used
prop.table(table(apply(motus.qual, 1, sum) > 0))
#>
#> FALSE TRUE
#> 0.8762552 0.1237448
# Corresponding proportion of artifactual reads (TRUE)
prop.table(xtabs(soil_euk$motus$count~apply(motus.qual, 1, sum) > 0))
#> apply(motus.qual, 1, sum) > 0
#> FALSE TRUE
#> 0.90582023 0.09417977
# Proportion of MOTUs and reads potentially artifactual for each criterion
apply(motus.qual, 2, sum) / nrow(motus.qual)
#> extraction_conta untargeted_taxon degraded_seq
#> 0.005218629 0.043567645 0.111567961
apply(motus.qual, 2, function(x) sum(soil_euk$motus$count[x])/sum(soil_euk$motus$count))
#> extraction_conta untargeted_taxon degraded_seq
#> 0.06669166 0.01372710 0.02629960
tmp.motus <-
apply(sapply(1:ncol(motus.qual), function(x) {
ifelse(motus.qual[,x]==T, colnames(motus.qual)[x], NA)}), 1, function(x) {
paste(sort(unique(x)), collapse = "|")
})
tmp.motus <- as.data.frame(gsub("^$", "not_artefactual", tmp.motus))
colnames(tmp.motus) <- "artefact_type"
ggplot(tmp.motus, aes(x=1, fill=artefact_type)) +
geom_bar() + xlim(0, 2) +
labs(fill="Artifact type") +
coord_polar(theta="y") + theme_void() +
scale_fill_brewer(palette = "Set3") +
theme(legend.direction = "vertical") +
ggtitle("MOTUs artefacts overview")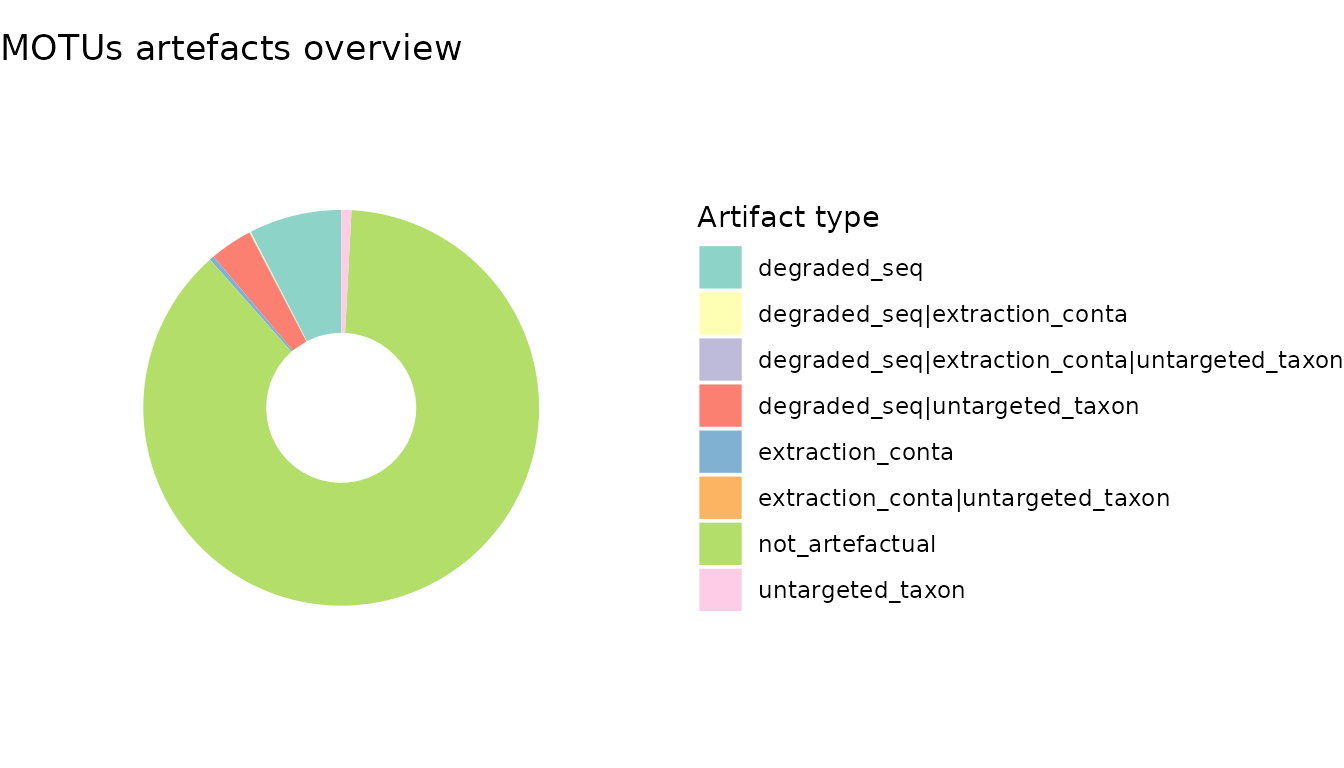
The above shows that MOTUs flagged as potentially artefactual account for ca. 10% of the dataset’s diversity and roughly the same in terms of readcount. Most of these artifact MOTUs are rare and correspond to sequences which are potentially highly degraded, with very low sequence similarity against the EMBL reference database. The most abundant artifacts MOTUs were identified as contaminants.
Let’s do the same with pcrs
# Create a table of pcrs quality criteria
# noise is identified as FALSE in soil_euk, the "!" transforms it to TRUE
pcrs.qual <- !soil_euk$pcrs[,c("low_contamination_level", "seqdepth_ok", "replicating_pcr")]
colnames(pcrs.qual) <- c("high_contamination_level", "low_seqdepth", "outliers")
# Proportion of pcrs potentially artifactual (TRUE) based on the criteria used
# excluding controls
prop.table(table(apply(pcrs.qual[soil_euk$pcrs$type=="sample",], 1, sum) > 0))
#>
#> FALSE TRUE
#> 0.8671875 0.1328125
# Proportion of MOTUs and reads potentially artifactual for each criterion
apply(pcrs.qual[soil_euk$pcrs$type=="sample",], 2, sum) / nrow(pcrs.qual[soil_euk$pcrs$type=="sample",])
#> high_contamination_level low_seqdepth outliers
#> 0.10937500 0.02734375 0.01562500
tmp.pcrs <-
apply(sapply(1:ncol(pcrs.qual), function(x) {
ifelse(pcrs.qual[soil_euk$pcrs$type=="sample",x]==T,
colnames(pcrs.qual)[x], NA)}), 1, function(x) {
paste(sort(unique(x)), collapse = "|")
})
tmp.pcrs <- as.data.frame(gsub("^$", "not_artefactual", tmp.pcrs))
colnames(tmp.pcrs) <- "artefact_type"
ggplot(tmp.pcrs, aes(x=1, fill=artefact_type)) +
geom_bar() + xlim(0, 2) +
labs(fill="Artifact type") +
coord_polar(theta="y") + theme_void() +
scale_fill_brewer(palette = "Set3") +
theme(legend.direction = "vertical") +
ggtitle("PCR artefacts overview")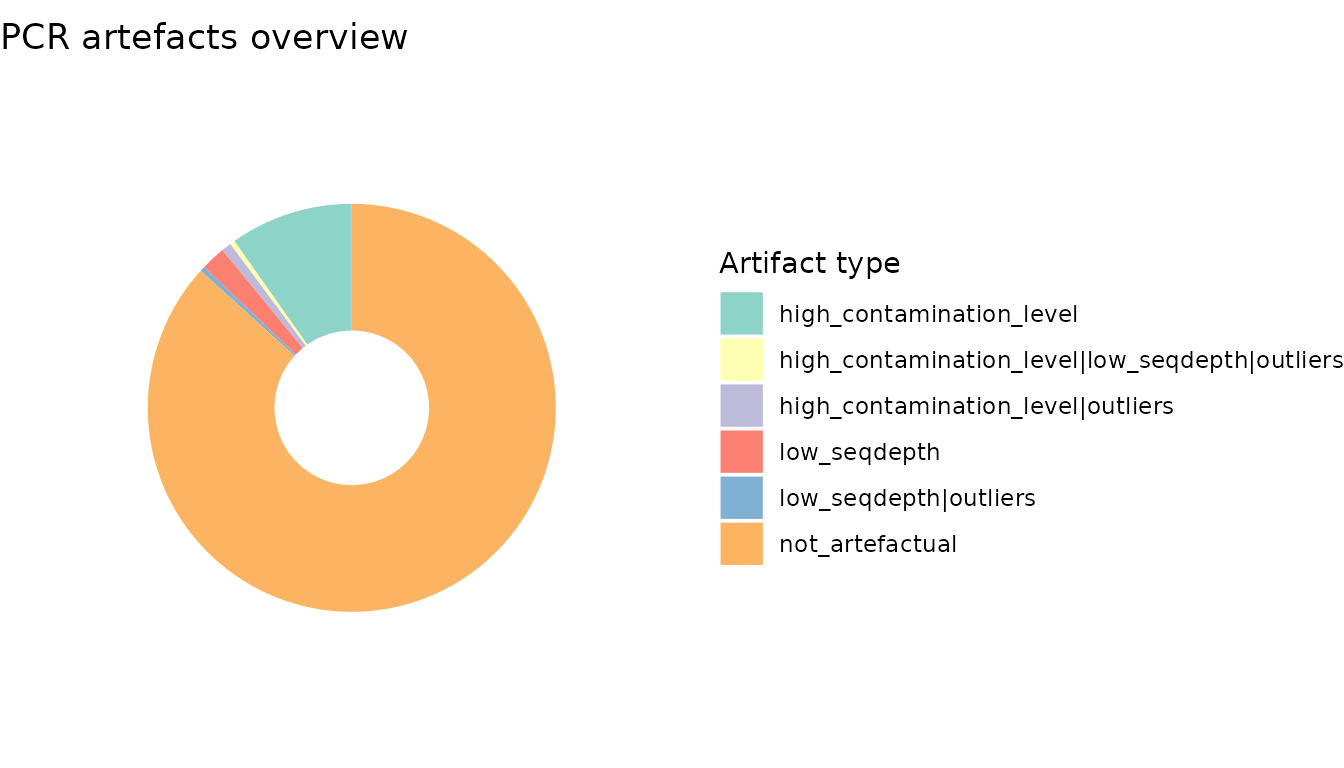
The pcrs flagged as potentially failed account for ca. 10% of the pcrs, most of these corresponding to pcrs yiedling a high amount of contaminants.
Data cleaning and aggregation
The final stage of the analysis consists in removing data based on the criteria defined above and aggregating pcrs to get rid of technical replicates. Here, the decision of which of these flagged criteria to use rests with the user, depending on how they feel the tagged errors impact on the characteristics of their dataset.
Removing spurious signal
First, we will remove suprious MOTUs, PCRs and adjusting read counts by removing tag-jumps . At this stage of the analysis, controls are no longer necessary, and so can also be removed from the dataset.
# Use tag-jump corrected metabarlist with the threshold identified above
tmp <- tests[["t_0.01"]]
# Subset on MOTUs: we keep motus that are defined as TRUE following the
# three criteria below (sum of three TRUE is equal to 3 with the rowSums function)
tmp <- subset_metabarlist(tmp, "motus",
indices = rowSums(tmp$motus[,c("not_an_extraction_conta", "target_taxon",
"not_degraded")]) == 3)
summary_metabarlist(tmp)
#> $dataset_dimension
#> n_row n_col
#> reads 384 11082
#> motus 11082 18
#> pcrs 384 16
#> samples 64 8
#>
#> $dataset_statistics
#> nb_reads nb_motus avg_reads sd_reads avg_motus sd_motus
#> pcrs 3173438 11082 8264.161 9543.057 285.3333 266.7837
#> samples 2514362 10886 9821.727 9453.080 420.4492 227.2386Same with pcrs
# Subset on pcrs and keep only controls
soil_euk_clean <- subset_metabarlist(tmp, "pcrs",
indices = rowSums(tmp$pcrs[,c("low_contamination_level",
"seqdepth_ok", "replicating_pcr")]) == 3 &
tmp$pcrs$type == "sample")
summary_metabarlist(soil_euk_clean)
#> $dataset_dimension
#> n_row n_col
#> reads 222 10570
#> motus 10570 18
#> pcrs 222 16
#> samples 61 8
#>
#> $dataset_statistics
#> nb_reads nb_motus avg_reads sd_reads avg_motus sd_motus
#> pcrs 2193430 10570 9880.315 9540.84 446.3333 228.1212
#> samples 2193430 10570 9880.315 9540.84 446.3333 228.1212Now check if previous subsetting leads to any empty pcrs or MOTUs
if(sum(colSums(soil_euk_clean$reads)==0)>0){print("empty motus present")}
if(sum(rowSums(soil_euk_clean$reads)==0)>0){print("empty pcrs present")}Since we have now removed certain MOTUs or reduced their read counts, we need to update some parameters in the metabarlist (e.g. counts, etc.).
soil_euk_clean$motus$count = colSums(soil_euk_clean$reads)
soil_euk_clean$pcrs$nb_reads_postmetabaR = rowSums(soil_euk_clean$reads)
soil_euk_clean$pcrs$nb_motus_postmetabaR = rowSums(ifelse(soil_euk_clean$reads>0, T, F))We can now compare some basic characteristics of the
soil_euk before and after data curation with
metabaR.
check <- melt(soil_euk_clean$pcrs[,c("nb_reads", "nb_reads_postmetabaR",
"nb_motus", "nb_motus_postmetabaR")])
check$type <- ifelse(grepl("motus", check$variable), "richness", "abundance")
ggplot(data = check, aes(x = variable, y = value)) +
geom_boxplot( color = "darkgrey") +
geom_jitter(alpha=0.1, color = "darkgrey") +
theme_bw() +
facet_wrap(~type, scales = "free", ncol = 5) +
theme(axis.text.x = element_text(angle=45, h=1))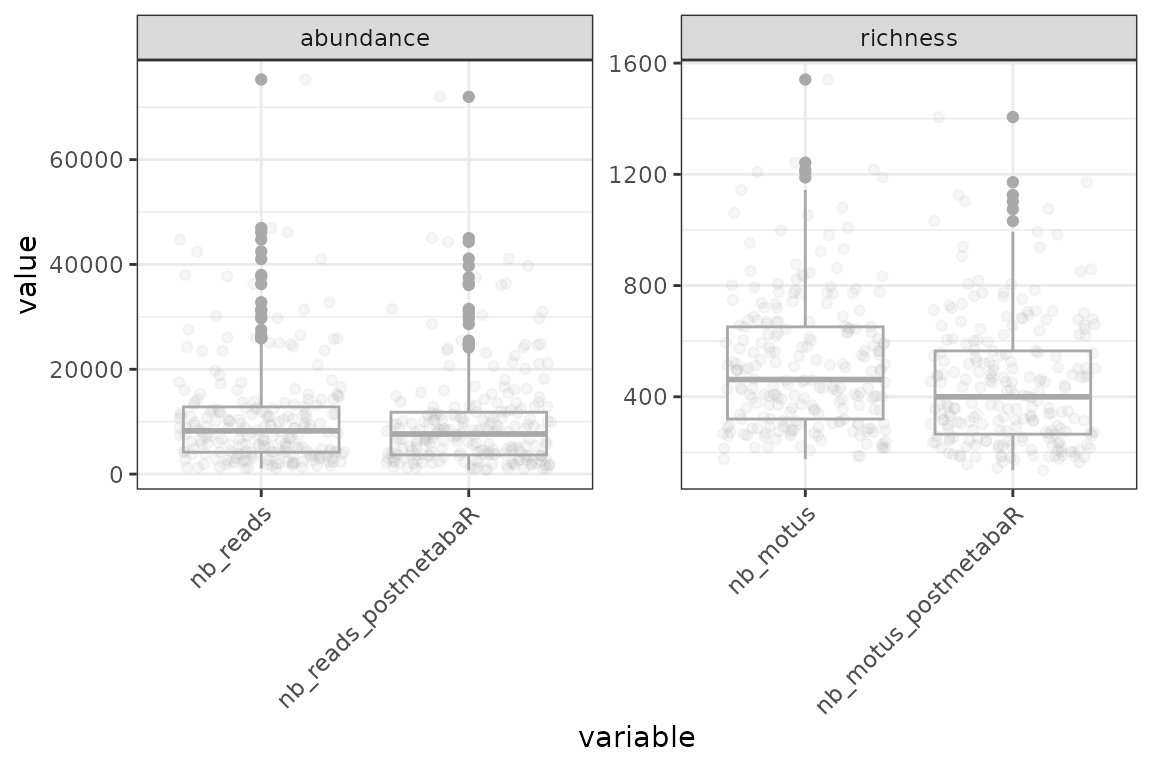
The sequencing depth and richness of pcrs are not greatly
affected by the trimming.
Let’s now see if the signal is changed in terms of beta diversity:
# Get row data only for samples
tmp <- subset_metabarlist(soil_euk, table = "pcrs",
indices = soil_euk$pcrs$type == "sample")
# Add sample biological information for checks
tmp$pcrs$Material <- tmp$samples$Material[match(tmp$pcrs$sample_id, rownames(tmp$samples))]
tmp$pcrs$Habitat <- tmp$samples$Habitat[match(tmp$pcrs$sample_id, rownames(tmp$samples))]
soil_euk_clean$pcrs$Material <-
soil_euk_clean$samples$Material[match(soil_euk_clean$pcrs$sample_id,
rownames(soil_euk_clean$samples))]
soil_euk_clean$pcrs$Habitat <-
soil_euk_clean$samples$Habitat[match(soil_euk_clean$pcrs$sample_id,
rownames(soil_euk_clean$samples))]
# Build PCoA ordinations
mds1 <- check_pcr_repl(tmp,
groups = paste(tmp$pcrs$Habitat, tmp$pcrs$Material, sep = " | "))
mds2 <- check_pcr_repl(soil_euk_clean,
groups = paste(
soil_euk_clean$pcrs$Habitat,
soil_euk_clean$pcrs$Material,
sep = " | "))
# Custom colors
a <- mds1 + labs(color = "Habitat | Material") +
scale_color_manual(values = c("brown4", "brown1", "goldenrod4", "goldenrod1")) +
theme(legend.position = "none") +
ggtitle("Raw data")
b <- mds2 + labs(color = "Habitat | Material") +
scale_color_manual(values = c("brown4", "brown1", "goldenrod4", "goldenrod1")) +
ggtitle("Clean data")
# Assemble plots
leg <- get_legend(b + guides(shape=F) +
theme(legend.position = "right",
legend.direction = "vertical"))
ggdraw() +
draw_plot(a, x=0, y=0, width = 0.4, height = 1) +
draw_plot(b + guides(color=F, shape=F), x=0.42, y=0, width = 0.4, height = 1) +
draw_grob(leg, x=0.4, y=0)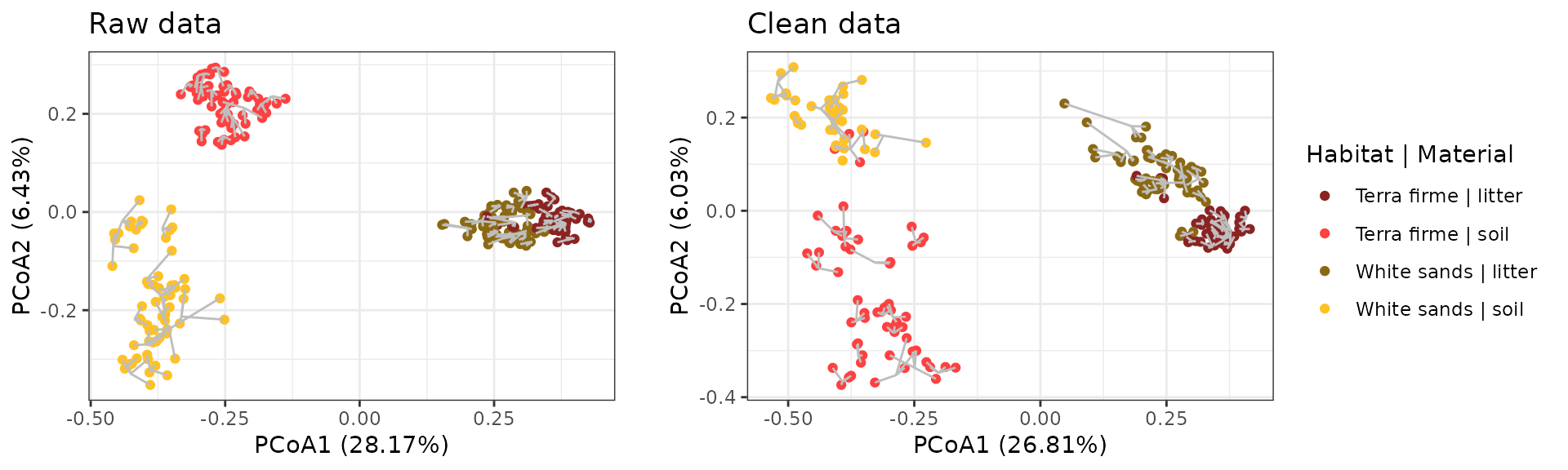
The resulting figure demonstrates that removing the various contaminants flagged above also increases the dissimilarities of samples both within and between- habitat and material types.
Data aggregation
At this stage, technical replicates of pcrs should be
aggregated so as to focus on biological patterns. This can be done with
the function aggregate_pcrs, which includes flexible
possibilities for data aggregation methods. Here, we will simply sum
MOTUs counts across pcrs replicates, which is the default
function of aggregate_pcrs. Note that this function can
also be used for other purposes than for aggregating technical
replicates. For example, if technical replicates are not included in the
experiment, one could instead aggregate biological replicates from a
same site.
soil_euk_agg <- aggregate_pcrs(soil_euk_clean)
summary_metabarlist(soil_euk_agg)
#> $dataset_dimension
#> n_row n_col
#> reads 61 10570
#> motus 10570 18
#> pcrs 61 20
#> samples 61 8
#>
#> $dataset_statistics
#> nb_reads nb_motus avg_reads sd_reads avg_motus sd_motus
#> pcrs 2193430 10570 35957.87 21718.55 965.0328 401.6088
#> samples 2193430 10570 35957.87 21718.55 965.0328 401.6088Now, soil_euk_agg has the same dataset characteristics
for pcrs and samples.
Final visualisations
Our data is now ready for ecological analyses with other
R packages. Before finishing this tutorial, we will
demonstrate the use of two last metabaR functions in
addition to those of other Rpackages to roughly explore the
characteristics of the final dataset.
Taxonomic composition
One challenge when assessing the taxonomic composition of DNA
metabarcoding data is (i) taxonomic information that is not always
available at the same taxonomic resolution, and (ii) the large taxonomic
breadth of the community under study. The ggtaxplot
function builds a taxonomic tree from information contained in the
motus table, in order to provide an overview of the
composition of the whole community:
ggtaxplot(soil_euk_agg,
taxo = "path",
sep.level = ":", sep.info = "@")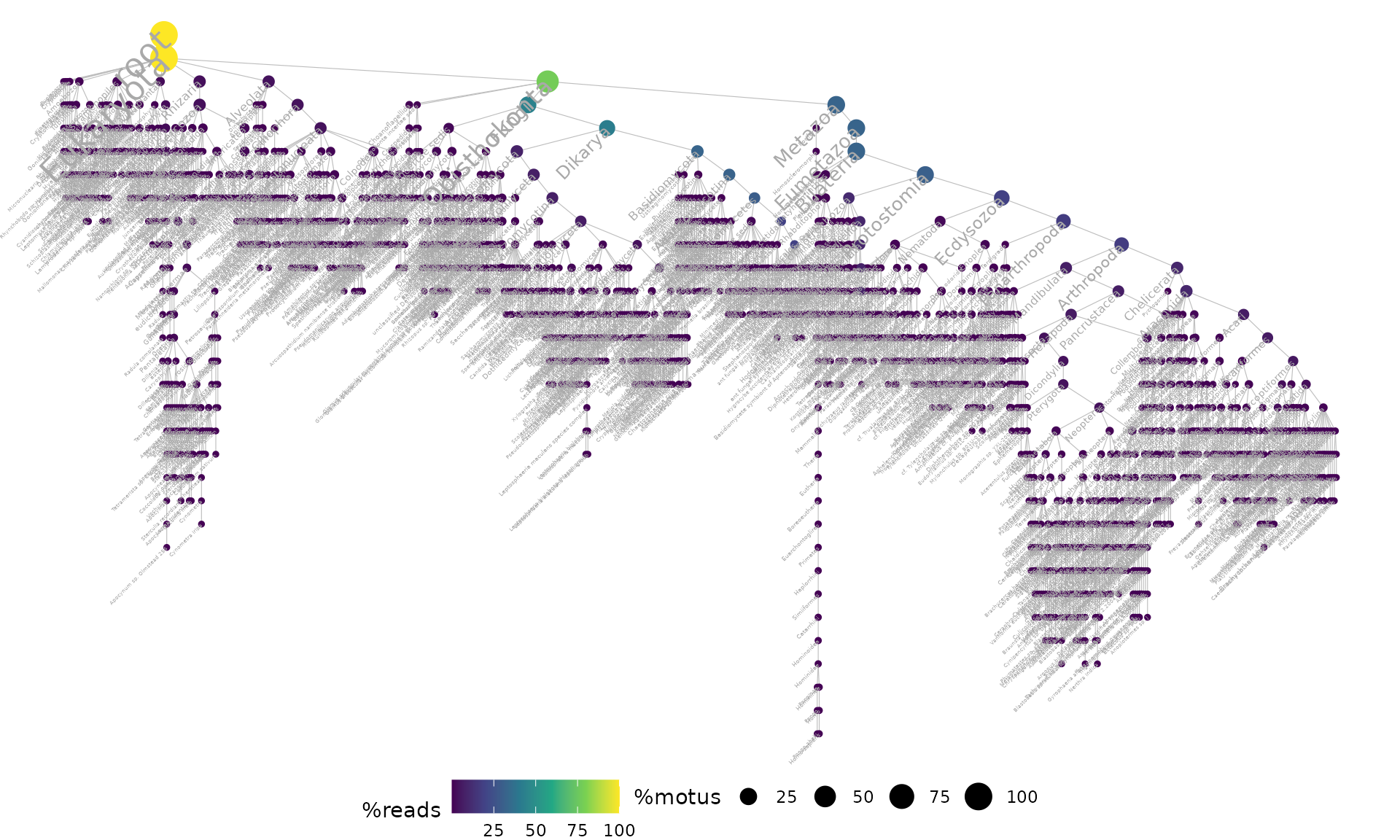
The taxonomic tree above shows that the soil_euk dataset
is mainy composed of Fungi and Metazoa MOTUs and reads. Let’s have a
further look at the taxonomic composition of phyla in one kingdom, the
Metazoa. This can be done with function aggregate_motus.
The kingdom level is not available as a single column in the
soil_euk dataset, and so requires the parsing of the full
MOTU taxonomic information available in the column “path”, with the
taxoparser function.
# Get the kingdom names of MOTUs
kingdom <-
unname(sapply(taxoparser(taxopath = soil_euk_agg$motus$path,
sep.level = ":",sep.info = "@"),
function(x) x["kingdom"]))
# Create metazoa metabarlist
soil_metazoa <- subset_metabarlist(soil_euk_agg, "motus",
kingdom=="Metazoa" & !is.na(kingdom))
# Aggregate the metabarlist at the phylum level
soil_metazoa_phy <-
aggregate_motus(soil_metazoa, groups = soil_metazoa$motus$phylum_name)
# Aggregate per Habitat/material
tmp <-
melt(aggregate(soil_metazoa_phy$reads, by = list(
paste(soil_metazoa_phy$samples$Habitat,
soil_metazoa_phy$samples$Material,
sep = " | ")), sum))
# Plot
ggplot(tmp, aes(x=Group.1, y=value, fill=variable)) +
geom_bar(stat="identity") +
labs(x=NULL, y="#reads", fill="Phyla") +
coord_flip() + theme_bw() +
scale_fill_brewer(palette = "Set3") +
theme(legend.position = "bottom") +
ggtitle("Metazoa phyla")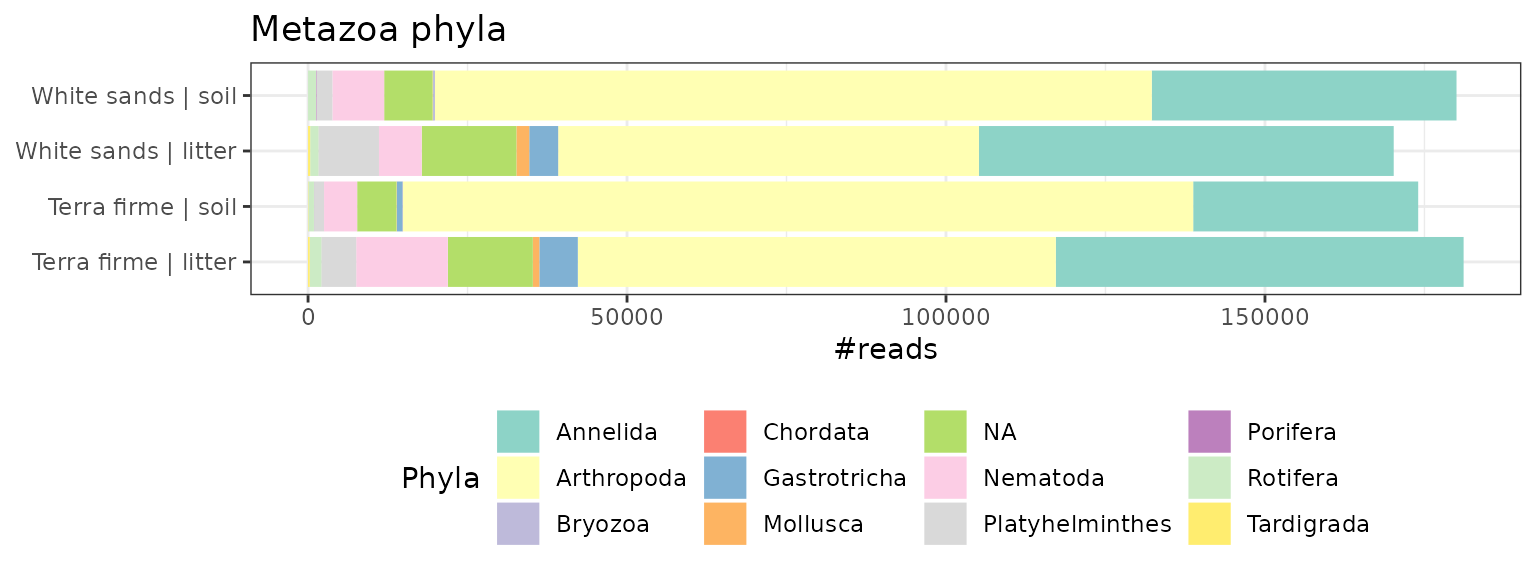
By considering a subset of our dataset, and aggregating at specific taxonomic levels, this approach has allowed us to uncover some initial trends in our data (e.g. that arthropod reads are higher in our soil samples versus our leaf litter samples).
Rarefaction curves
To finish, lets take a final look at the diversity coverage of samples with rarefaction curves (rather than our previous look at the pcrs level).
soil_euk_agg.raref = hill_rarefaction(soil_euk_agg, nboot = 20, nsteps = 10)
material <- paste(soil_euk_agg$samples$Habitat, soil_euk_agg$samples$Material)
material <- setNames(material,rownames(soil_euk_agg$samples))
#plot
p <- gghill_rarefaction(soil_euk_agg.raref, group=material)
p + scale_fill_manual(values = c("brown4", "brown1", "goldenrod4", "goldenrod1")) +
scale_color_manual(values = c("brown4", "brown1", "goldenrod4", "goldenrod1")) +
labs(color="Habitat | Material type")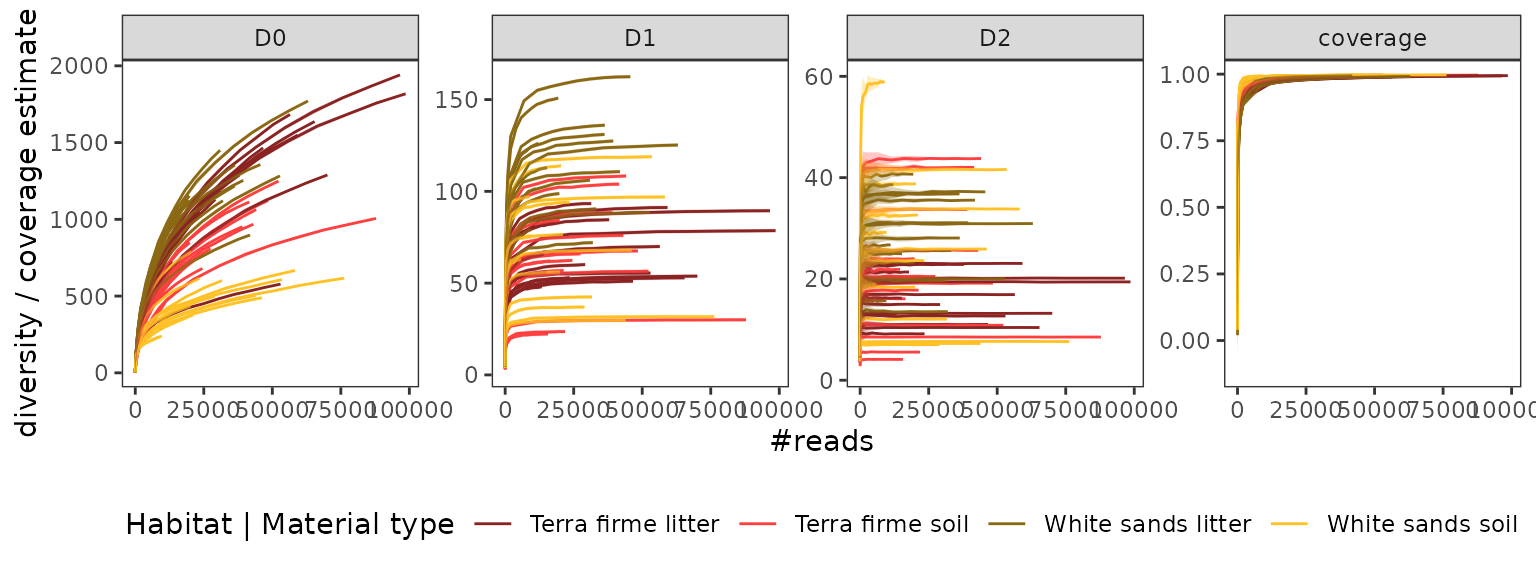
This visualisation allows us to explore patterns of diversity across
our sample substrates and habitats. You can now go on with ecological
analyses using metabaR for data handling and any other R
package!
Hopefully this tutorial has demonstrated the strength of
metabaR in visualising data, its use for identifying and
removing surious sequences, and ultimately its complementary nature with
existing packages for data handling. We hope that you are encouraged to
incorporate the above ideas into your future processing of metabarcoding
data!