
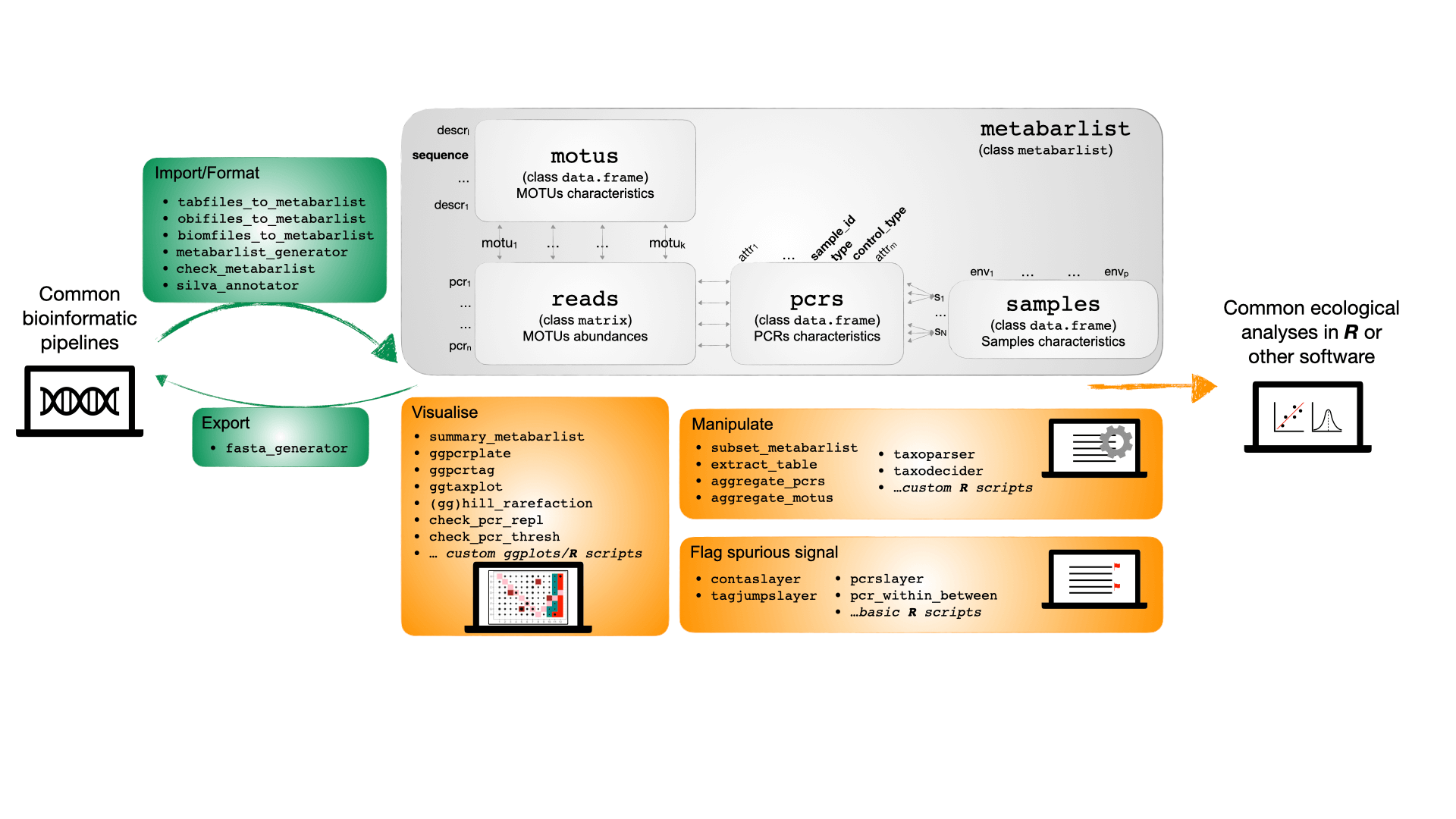
metabaR is an R package enabling the import, handling and processing of DNA metabarcoding data that have been already processed through bioinformatic pipelines. It provides functions to reveal and filter common molecular artifacts produced during the experimental workflow.
This package can be easily used in combination with others R packages commonly used in ecology (vegan, ade4, ape, picante, etc.), and provides flexible graphic systems based on ggplot2 to visualise the data under both ecological and experimental perspectives.
More specifically, metabaR provides:
- Import functions of DNA metabarcoding data from different bioinformatics pipelines
- Functions to manipulate the different types of tables one usually deals with when working with DNA metabarcoding.
- Functions of data curation that are absent from the above pipelines and detect/flag potential molecular artifacts such as contaminants, dysfynctional PCRs, etc.
- Functions to visualise the data under both ecological (e.g. type of samples, rarefaction curves) and experimental (e.g. type of controls, distribution across the PCR plate design) perspectives.
metabaR is developed on GitHub:
https://github.com/metabaRfactory/metabaR
Installation
metabaR can be installed from GitHub using:
# install bioconductor dependencies
install.packages("BiocManager")
BiocManager::install("biomformat")
# install metabaR package
install.packages("remotes")
remotes::install_github("metabaRfactory/metabaR")Package dependencies: - for graphical purposes: igraph, ggplot2 and cowplot - for formatting purposes: reshape2, seqinr, biomformat - for analysis purposes: vegan, ade4
Example
This is a basic example of use:
library(metabaR)
data(soil_euk)
summary_metabarlist(soil_euk)
#> $dataset_dimension
#> n_row n_col
#> reads 384 12647
#> motus 12647 15
#> pcrs 384 11
#> samples 64 8
#>
#> $dataset_statistics
#> nb_reads nb_motus avg_reads sd_reads avg_motus sd_motus
#> pcrs 3538913 12647 9215.919 10283.45 333.6849 295.440
#> samples 2797294 12382 10926.930 10346.66 489.5117 239.685Citation
Zinger, L., Lionnet, C., Benoiston, A.‐S., Donald, J., Mercier, C. and Boyer, F. (2021), Metabar: an R package for the evaluation and improvement of DNA metabarcoding data quality. Methods Ecol Evol. https://doi.org/10.1111/2041-210X.13552